










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de la Sociedad Química de México
Print version ISSN 0583-7693
Rev. Soc. Quím. Méx vol.44 n.1 Ciudad de México Jan./Mar. 2000
Investigación
Asignación inequívoca de las señales del espectro de resonancia magnética nuclear de 1H y 13C de monoterpenos empleando métodos computacionales
Fernando Cortés,1 Gabriel Cuevas,*1, Julieta Tenorio2 y Ana L. Rochín2
1 Instituto de Química, Universidad Nacional Autónoma de México, Cd. Universitaria, Apdo. Postal 70213, 04510 Coyoacán México, D.F. México.
2 Escuela de Ciencias Químicas, Universidad La Salle. Benjamín Franklin 47, 06140, México, D.F. México.
Recibido el 14 de diciembre de 1999.
Aceptado el 2 de febrero del 2000.
Resumen
Se determinaron los corrimientos químicos tanto de 1H como de 13C del canfeno y el α-pineno usando el método que permite la Suma Sobre los Estados incluyendo perturbaciones en la Teoría de Funcionales de la Densidad utilizando el programa modificado deMon-KS. Los cálculos no solo reproducen cuantitativamente los corrimientos químicos observados experimentalmente para el núcleo de 1H y cualitativamente para el de caso de 13C, sino que permiten asignar inequívocamente las señales de los espectros correspondientes.
Palabras clave: RMN, DFT, canfeno, pineno, química computacional.
Abstract.
Ab initio calculations, within the frame of Density Functional Theory were carried out on camphene and α-pinene. The 1H and 13C NMR shifts were estimated according to the recently developed Sum-Over-States Density Functional Perturbation Theory (SOS-DFPT) as implemented in a modified deMon-KS program. The calculations not only reproduced the observed NMR chemical shifts, quantitatively in the case of 1H nuclei and qualitatively in the case of 13C nuclei, but also allow assigning unambiguously the signal on these spectra.
Keywords: RMN, DFT, camphene, pinene, computational chemistry.
A la memoria del Dr. Raúl Cetina Rosado
Introducción
La asignación de las señales de los espectros de Resonancia Magnética Nuclear (RMN) de 1H y de 13C de hidrocarburos es difícil y ha llevado a errores que han requerido múltiples revisiones.
El desarrollo de la química computacional, tanto en el aspecto del cómputo mismo (procesadores más rápidos, recursos más grandes de memoria, cómputo en paralelo, etc.) así como de la química teórica (nuevas teorías, conceptos innovadores, nuevos algoritmos) y de las matemáticas (métodos numéricos más eficientes, por ejemplo), permiten actualmente el cálculo de las propiedades electrónicas de las moléculas que se estudian experimentalmente, de tal modo que es posible obtener propiedades termodinámicas y espectroscópicas [1] más confiables de las mismas y no sólo de modelos, en ocasiones sobre simplificados.
La combinación del cálculo de los desplazamientos químicos por métodos a primeros principios (ab initio) y los datos experimentales de resonancia magnética nuclear, proveen una herramienta que puede ser aplicada rutinariamente para la elucidación y caracterización de nuevos compuestos. Sin embargo su uso no es generalizado y las principales aplicaciones se han presentado en el estudio de iones carbenio [2] y moléculas en donde los efectos estereoelectrónicos son importantes [3] debido a que hasta hace poco las moléculas orgánicas "pequeñas" eran incalculables con niveles de teoría aceptables. Además de que estas predicciones necesitan llevarse a cabo a un costo computacional razonable.
El cálculo de los desplazamientos químicos de 1H y de 13C se ha efectuado por varios métodos y utilizando diferentes bases, para resolver el problema ocasionado por la anisotropía propia del corrimiento químico, como LORG (Orbitales localizados, origen local), IGLO (Orbitales localizados gauge individuales) [4] o GIAO (Orbitales atómicos gauge incluidos) [5] que se han aplicado a la asignación de señales en moléculas pequeñas. También se ha realizado cálculos sobre la estructura del Taxol [6] y algunos terpenos [7] pero con una diferencia significativa con los resultados experimentales, ya que el tamaño de la molécula y las limitaciones computacionales obligaron a emplear niveles bajos de teoría.
En este trabajo se presentan los resultados del cálculo de los desplazamientos químicos de 1H y de 13C de dos hidrocarburos, obtenidos por el método de suma sobre los estados empleando perturbaciones en la teoría de funcionales de la densidad con el fin de ilustrar la utilidad de la química computacional en la asignación de señales en resonancia magnética nuclear. El canfeno (1) y el α-pineno (2) son los ejemplos aquí analizados, elegidos por el número de trabajos experimentales reportados sobre la asignación de sus señales en resonancia magnética nuclear (Fig. 1). Es importante destacar que la ausencia de grupos funcionales polares en este tipo de moléculas dificulta su asignación experimental.

Métodos computacionales
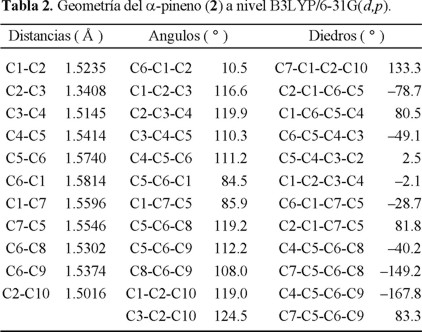
La optimización completa de la geometría de las moléculas aquí estudiadas fue realizada usando la teoría de funcionales de la densidad (TFD). La energía semilocal de intercambio-correlación usada fue la incluida en el funcional Becke3LYP [8]. Se empleó una base doble ξ de valencia dividida que incluye polarización tanto en átomos pesados como ligeros; la 6-31G(d,p). La optimización completa de la geometría fue realizada con el programa Gaussian 94 y en las tablas 1 y 2 se muestran los datos geométricos de las moléculas así optimizadas.

Los desplazamientos químicos de 1H y de 13C fueron calculados de acuerdo con la teoría de las perturbaciones de funcionales de la densidad con la suma sobre los estados como se implementa en el programa modificado de deMonKS [9]. Para los desplazamientos químicos se usaron la energía de intercambio semilocal de Perdew-Wang [10] y la función de correlación de Perdew (PP) [11]. La base utilizada en el cálculo de los desplazamientos fue la IGLOIII de Kutzelnigg [12].
Los desplazamientos químicos calculados en este trabajo son relativos al tetrametilsilano (TMS) y sus desplazamientos fueron obtenidos en el nivel teoría PP/IGLOIII//B3LYP/6-31G(d,p ) con un valor de la constante de desprotección para el 13C de 179.15 y para el 1H de 31.275 [13].
Resultados y discusión
A pesar que los espectros de RMN 13C de ambos compuestos aparecen con regularidad en la literatura, varias de sus asignaciones son equívocas, controversiales o ambiguas. En las tablas 3 y 4 se muestran algunas de las asignaciones reportadas en varios trabajos realizados por Grutzner [14], Werstiuk [15], Morris [16], Grover [17], y Brunke [18], para canfeno (1) y por Brun [19] para el α-pineno. Para el caso del canfeno (1) la ambigüedad se presenta en la asignación de los metilos 8 y 9 mientras que para el caso del α-pineno (2) lo ha sido la asignación de los carbonos 4 y 7. Offerman [20] desarrolló un trabajo exhaustivo para asignar las señales de 13C empleando análisis gráfico de series de frecuencias en espectros desacoplados fuera de resonancia, acoplamiento C-H a una enlace y a larga distancia en espectros desacoplados, corrimientos inducidos por plata/lantanidos, separación quiral y desplazamientos calculados empíricamente. Estos datos fueron tomados como referencia experimental para este trabajo ya que su trabajo es el más reciente, completo y riguroso.

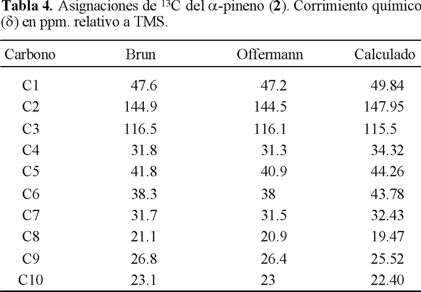
Los cálculos realizados a nivel PP/IGLOIII//B3LYP/6-31(d,p) dan como resultado desplazamientos químicos que están cercanos a los obtenidos por Offerman tanto para el canfeno como para el α-pineno, esto se muestra en las tablas 3 y 4, pero se presenta de manera clara en las figuras 2 y 3, donde se observa que los datos calculados siguen la tendencia de los experimentales, lo que permite contribuir a la asignación inequívoca.


Los valores calculados corresponden al valor del tensor de protección anisotrópica que no es el valor que reportan los resultados de RMN experimental ya que se requiere una referencia interna arbitraria que suele ser TMS. Para poder comparar los resultados teóricos y experimentales es necesario incorporar una primera corrección que implica que los desplazamientos aquí calculados fueran referidos al desplazamiento químico del TMS, calculado en el mismo nivel de teoría. En segundo lugar las moléculas se calculan considerándolas aisladas, a 0 K y en fase vapor, por lo que resulta sorpredente la proximidad entre los resultados teóricos y los experimentales, sin embargo para poder efectuar una mejor comparación es importante reducir la influencia de estas condiciones, lo que se hace al incorporar una ecuación empírica. Por lo que los desplazamientos fueron corregidos por la ecuación empírica y = 1.057 x + 2.12. Como se observa en la Fig. 4 se encuentra una tendencia lineal que habla de la proximidad de los desplazamientos calculados a los experimentales.

Con respecto a los carbonos cuya asignación resulta difícil, esta metodología resulta de utilidad, ya que con los desplazamientos calculados para los carbonos 8 y 9 del canfeno y 4 y 7 del α-pineno es posible efectuar la asignación inequívoca. A pesar de que la diferencia entre el valor experimental y el valor calculado es de 0.6 ppm en promedio para el canfeno y 2 ppm en promedio para el pineno, se obtienen desplazamientos calculados para cada carbono que permite distinguir a cada uno en cada par problema, con la ventaja de que cada carbono se encuentra definido desde el principio del cálculo, cuando se construye la geometría de partida, mientras que en el procedimiento experimental sólo se cuenta con el valor del desplazamiento. El cálculo puede tener un determinado error respecto al valor experimental pero la tendencia del corrimiento químico se preserva de modo tal que el orden de asignación no puede variar.
De la misma manera se puede calcular el desplazamiento químico de 1H del canfeno (1) y del α-pineno (2). Los desplazamientos referidos a tetrametilsilano se muestran en la Tabla 5.

Si se comparan los desplazamientos de H calculados con el espectro de RMN 1H se encuentra que los datos calculados están muy cercanos a los experimentales, lo necesario para poder elucidar el espectro.
Con esto se demuestra la capacidad de los métodos de cálculo rutinarios y su utilidad para determinar los desplazamientos químicos con cálculos ab-initio a nivel PP/IGLOI-II//B3LYP/6-31(d,p) para la asignación inequívoca de señales espectrales en resonancia magnética nuclear.

Agradecimientos
Agradecemos a DGSCA de la UNAM y a la Coordinación de Sistemas de la Universidad La Salle por el tiempo de computo proporcionado y a DGAPA (Proyecto IN107597) y al CONACyT (Proyecto 32420E) por el financiamiento parcial de este proyecto así como las becas otorgadas a Julieta Tenorio y Ana Rochín.
Referencias bibliográficas
1. Ziegler, T. Chem. Rev. 1991, 91, 651-667. [ Links ]
2. Saunders: M.; Laidig, K. E.; Wolfsberg, M. J. Am. Chem. Soc. 1989, 111, 8989-8994. [ Links ]
3. Cuevas, G.; Juaristi, E.; Vela, A. J. Phys. Chem. 1999, 103, 932-937. [ Links ]
4. Cuevas, G.; Juaristi, E.; Vela, A. J. Mol. Struct. (Teochem) 1997, 418, 231-241. [ Links ]
5. Bühl, M., Kaupp, M., Malkina, O. L., Malkin, V. G., J. Computational Chem., 1999, 20, 91-105. [ Links ]
6. Cheeseman, J. R.; Trucks, G. W.; Keith, T. A.; Frish M. J. J. Chem. Phys. 1996, 104, 5497-5509. [ Links ]
7. Forsyth D. A.; Sebag A. B. J. Am. Chem. Soc. 1997, 119, 9483-9494. [ Links ]
8. Becke, A. D. J. Chem. Phys. 1988, 88, 1053-1062. [ Links ] Becke A. D. Phys. Rev. A 1988, 38, 3098-3100. [ Links ]
9. St-Amant, A.; Salahub, D.R.; Chem. Phys. Lett. 1990, 169, 387-392; [ Links ] Salahub, D.R.; Castro, M. E.; Proynov, E. I., en: G. L. Malli (Ed.), Relativistic and Electron Correlation Effects in Molecules and Solids, Vol. 318 of NATO ASI Sereis B: Physics, Plenum Press, New York, 1994, p. 411; [ Links ] Salahub, D.R.; Castro, M. E.; Fournier, R.; Calaminici, P.; Godbout, N.; Goursot, A.; Jamorski, C.; Kobayashi, H.; Martínez, A.; Papai, I.; Proynov, E.; Russo, N.; Sirois, S.; Ushio, J.; Vela, A. en: H. Sellers, J. T. Golab (Eds.), Theoretical and Computational Approaches to interfase Phenomena, Plenum Press, New York, 1995, p. 187. [ Links ]
10. Perdew, J. P.; Wang, Y. Phys. Rev. B, 1986, 33, 8800-8802. [ Links ]
11. Perdew, J. P. Phys. Rev. B, 1986, 33, 8822-8824; [ Links ] Perdew, J. P., Phys. Rev. B, 1986, 34, 7406. [ Links ]
12. Kultzelnigg, W.; Fleischer, U.; Schindler, M. en: NMR-Basic Principles and Progress, Vol. 33, Springer-Verlag, Heidelberg. 1990, p. 165. [ Links ]
13. Referencia 4.
14. Grutzner, J. B.; Jautelat, M.; Dence, J. B.; Smith, R. A.; Roberts, D. J. Am. Chem. Soc. 1970, 7107-7120. [ Links ]
15. Werstiuk, N. H.; Taillefer, R.; Bell, R. A. Can. J. Chem. 1972, 50, 2146-2149. [ Links ]
16. Morris, D. G.; Murray, A. M., J. Chem. Soc. Perkin Trans. 2 1975, 539-541. [ Links ]
17. Grover, S. H.,; Stothers, J. B. Can. J. Chem. 1975, 53, 589-595. [ Links ]
18. Brunke, J.; Hammerschmidt, F. J. Tetrahedron Lett. 1980, 2405-2408. [ Links ]
19. Brun, P.; Casanova, J.; Hatem, J. Org. Magn. Reson. 1979, 12, 537-541. [ Links ]
20. Offermann, W. Org. Magn. Reson. 1982, 20, 203-204. [ Links ]

