











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El desarrollo de fármacos es un proceso complejo el cual inicia con la identificación de compuestos que se unen a un blanco terapéutico o que muestran actividad biológica en un ensayo de tamizaje. Aquellas moléculas que muestran actividad biológica son llamadas hits. El siguiente paso es encontrar compuestos que tengan propiedades farmacéuticas atractivas, incluyendo baja toxicidad, solubilidad acuosa adecuada para administrarse vía oral, entre otras propiedades farmacocinéticas. Tales compuestos son llamados «líderes o cabezas de serie». Típicamente, los hits son encontrados por tamizaje de un número vasto de moléculas, mientras que los compuestos «cabezas de serie» son desarrollados a partir de los hits a través de modificaciones químicas. Considerando que el número de moléculas orgánicas que son sintéticamente factibles se encuentran entre 1020 y 1024 (Ertl, 2003), es evidente que su análisis sería muy complejo sin el uso de técnicas computacionales.
En este artículo se discuten métodos computacionales comunes que se emplean durante el desarrollo de fármacos. El objetivo es contribuir a la enseñanza de Química Farmacéutica. Trabajos relacionados han sido publicados en la revista Educación Química; a diferencia del trabajo de Medina-Franco, Lopez-Vallejo y Castillo (2006) publicado hace 10 años, este artículo enfatiza la aplicación de métodos computacionales en el contexto de conceptos novedosos que han surgido en el diseño de fármacos. Aplicaciones específicas al desarrollo de medicamentos dirigidos a dianas epigenéticas se encuentran publicadas en Medina-Franco, Méndez-Lucio, Yoo y Dueñas, (2015). Se hace notar que el enfoque del artículo publicado en 2015 está centrado en 4 líneas de investigación específicas y casos de estudio particulares.
Diseño y desarrollo de fármacos
La mayoría de los medicamentos que están en uso clínico son el resultado de un proceso de investigación muy complejo. Por lo mismo, es necesaria la unión de esfuerzos de diferentes disciplinas científicas para descubrir y desarrollar medicamentos con efectos clínicos benéficos y efectos secundarios mínimos. Aunque el descubrimiento y desarrollo de medicamentos se ha hecho durante muchos años usando únicamente métodos experimentales, se espera que el proceso se acelere gracias al uso de métodos de cómputo (también llamados in silico) que permiten codificar con precisión modelos teóricos y son capaces de procesar grandes cantidades de información. Además, en muchos proyectos, la aplicación de modelos generados in silico contribuye a entender los mecanismos de acción de los principios activos de los medicamentos o a mejorar las propiedades de los mismos. Un ejemplo es la optimización de los fármacos de origen natural. En la actualidad se hacen esfuerzos para mejorar las propiedades de estos medicamentos (como reducir efectos secundarios) o buscar actividades biológicas potenciales de compuestos químicos aislados de fuentes naturales (Medina-Franco, 2013).
Etapas del desarrollo de fármacos
La Figura 1 muestra las etapas principales de un modelo clásico para el desarrollo de un medicamento. El proceso comienza con la investigación sobre las causas de una enfermedad, que en algunos casos puede llevar a la identificación de una o varias dianas moleculares asociadas con esa enfermedad. Los pasos siguientes involucran la identificación de compuestos activos con la diana molecular y la optimización de su actividad biológica. Estos ensayos se hacen in vitro con blancos moleculares aislados de las células. Los compuestos activos se someten a varias evaluaciones experimentales que implican ensayos en líneas celulares, en animales y pruebas clínicas en humanos. Los compuestos que pasan satisfactoriamente por todas las etapas son aprobados para uso clínico por un agente regulatorio, por ejemplo, la Comisión Federal para la Protección contra Riesgos Sanitarios (COFEPRIS) en México o la Food and Drug Administration (FDA) en Estados Unidos.

Probabilidades de éxito
En el desarrollo de fármacos, la mayoría de los compuestos que muestran actividades in vitro con las dianas moleculares fallan las pruebas siguientes. Esto se debe frecuentemente a sus pobres propiedades farmacocinéticas y toxicidad. Es decir, además de que un compuesto es activo con los blancos moleculares deseados, también afecta otros procesos fisiológicos y no pueden usarse en forma segura en humanos. Se estima que de cada 9,000 moléculas biológicamente activas solo una tiene uso clínico.
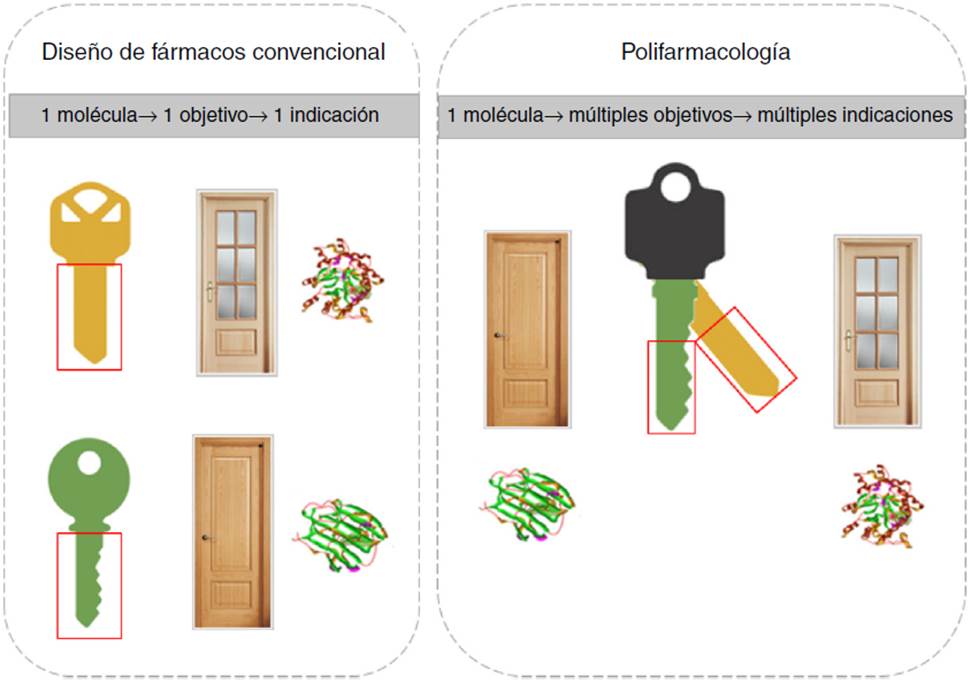
A pesar de que el modelo de desarrollo de fármacos de la Figura 1 se sigue aplicando con éxito, no necesariamente es el más eficiente. Cada vez hay mayor evidencia que un fármaco interacciona con diversos blancos moleculares. En varios casos el efecto clínico se debe a la interacción con múltiples dianas, dando origen al concepto de polifarmacología. De esta manera, la polifarmacología está modificando el paradigma actual de diseño de fármacos, de un diseño dirigido a una sola diana al diseño dirigido simultáneamente a múltiples blancos terapéuticos. Esta estrategia se conoce como diseño multi-objetivo y una de las metas es diseñar una «llave maestra» que se una selectivamente a una serie de blancos moleculares que produzcan una respuesta clínica deseada. La polifarmacología y el diseño de fármacos que actúan como «llaves maestras» se ilustran en la Figura 2.
Costo y tiempo aproximado
Se estima que el desarrollo de un fármaco tarda aproximadamente entre 10 y 15 años y se invierten en promedio 800 millones de dólares, teniendo el mayor costo y tiempo las pruebas clínicas en humanos. El tiempo y costos tan elevados están asociados en gran medida a la gran cantidad de moléculas que fallan una o varias etapas del desarrollo de fármacos.
Estrategias para identificar compuestos líderes
Hay diversas estrategias experimentales y/o computacionales para identificar compuestos activos que se consideran líderes para implementar un programa de optimización:
Optimización de fármacos ya aprobados para su uso clínico. La optimización puede consistir en mejorar la eficacia del medicamento o reducir efectos secundarios.
Ensayo biológico sistemático de colecciones de compuestos. Un caso típico son las pruebas biológicas de alto rendimiento (en inglés high-throughput screening [HTS]). En HTS se usan robots para evaluar en pocas horas colecciones con miles o millones de moléculas.
Uso de información biológica disponible. Por ejemplo, si se conoce el mecanismo de acción de un sustrato se diseñan compuestos que se espera sigan el mismo mecanismo de acción.
Diseño de fármacos asistido por computadora (DIFAC). Esta estrategia involucra una o varias técnicas que se discuten en la siguiente sección.
Diseño de fármacos asistido por computadora
El DIFAC tiene como principio entender las relaciones estructura-actividad biológica o farmacológica de compuestos (Medina-Franco et al., 2006).
Objetivos del DIFAC
Los objetivos del DIFAC pueden dividirse en 3:
Diseño o identificación de nuevos compuestos. Esto es, diseñar nuevas estructuras con efecto biológico en una categoría terapéutica deseada. Una alternativa es buscar dentro de colecciones de compuestos existentes moléculas que tengan una actividad biológica específica.
Seleccionar candidatos. Los métodos computacionales ayudan a distinguir las moléculas sobre las cuales se deben enfocar primero las pruebas experimentales, por ejemplo, compra, síntesis, y evaluación biológica. Sin embargo, los métodos de cómputo no reemplazan a los experimentos.
Optimizar líderes para mejorar las propiedades deseadas y disminuir los efectos adversos.
El DIFAC es una integración de métodos
El DIFAC está integrado por diversas áreas de la investigación que incluyen a la quimio informática, bioinformática, modelado molecular, química teórica y visualización de datos. Cada una comprende métodos que se emplean dependiendo de las características del sistema, información experimental disponible y de los objetivos específicos del proyecto.
Falsas expectativas del DIFAC
El concepto de «diseño de fármacos por computadora» está frecuentemente asociado con falsas expectativas. La Tabla 1 resume ejemplos los cuales se describen brevemente a continuación.
Tabla 1 Resumen de falsas expectativas típicas del DIFAC y visión realista
| Falsa expectativa | Visión realista |
|---|---|
| Las computadoras pueden diseñar fármacos | Los métodos computacionales no diseñan fármacos por sí solos; forman parte de un esfuerzo multidisciplinario para alcanzar un objetivo extremadamente complejo |
| Un cálculo teórico (por ejemplo de acoplamiento molecular o docking) demuestra la unión fármaco-blanco molecular | Cálculos de acoplamiento molecular dan hipótesis y proponen modelos de unión; se utilizan como apoyo a los experimentos que son los que demuestran la unión |
| Se puede diseñar fármacos apretando botones (push-a-bottom drug discovery) | El DIFAC es un proceso que requiere gran cantidad de prepración, análisis e interpretación de resultados que se obtienen de la computadora |
| El DIFAC es rápido porque lo hacen las computadoras | No; la prepración de datos e intepretación de resultados, que forman parte fundamental del DIFAC, son procesos que requieren de un tiempo que puede durar meses |
Una falsa expectativa es que las computadoras pueden diseñar medicamentos por sí solas. Esta percepción conduce con frecuencia a la pregunta: ¿algún medicamento se ha diseñado por métodos computacionales? Los métodos de cómputo no diseñan de forma automatizada los medicamentos. Para que sean efectivos, los modelos teóricos deben integrarse como pruebas biológicas o biofísicas. Los métodos de cómputo forman parte de un esfuerzo multidisciplinario que involucra otras áreas como síntesis orgánica, productos naturales y HTS. Por ejemplo, los cálculos computacionales no demuestran que una molécula puede unirse a una enzima. En la discusión de acoplamiento molecular automatizado se verá que los cálculos «asisten» en la predicción del modo de unión de una molécula con un blanco molecular, pero no demuestran dicha unión. Otras falsas expectativas son que se pueden diseñar fármacos «apretando botones» y que los análisis de cómputo son rápidos. Si es cierto que el procesamiento de datos es rápido (dependiendo del tipo de cálculo, equipo de cómputo y sistema en estudio), el análisis e interpretación de datos son laboriosos. En forma semejante a una prueba experimental, hay que validar los métodos computacionales y encontrar los parámetros óptimos para que los cálculos estén apegados a la realidad.
Métodos computacionales comunes
Como se mencionó arriba, el desarrollo de fármacos involucra varias etapas que abarcan desde la identificación de dianas moleculares hasta las fases clínicas (Fig. 1). La mayoría de las técnicas clásicas del DIFAC están centradas en las primeras etapas. En esta sección se discuten 3 métodos que se emplean con frecuencia en la identificación de compuestos líder y su optimización.
Acoplamiento molecular automatizado
El acoplamiento molecular automatizado (o en inglés molecular docking) consiste en buscar la conformación y posición óptima de un ligando (por ejemplo, de una molécula orgánica pequeña) dentro de una diana molecular (por ejemplo, una enzima, un canal iónico o un receptor acoplado a proteína G). La Figura 3 ilustra un modelo de acoplamiento de la isoforma BRD4 (bromodomain de la familia BET), una diana epigenética. Mediante búsquedas por similitud molecular se encontró que la pentoxifilina es un potencial inhibidor de BRD4; en púrpura se muestra la conformación del ligando propuesto, que es semejante a la orientación del ligando cocristalizado (A). ¿Cómo podemos confiar en el resultado? Para ello aplicamos el mismo algoritmo sobre el ligando cocristalizado, para comprobar si es capaz de recuperar el confórmero observado experimentalmente (B).
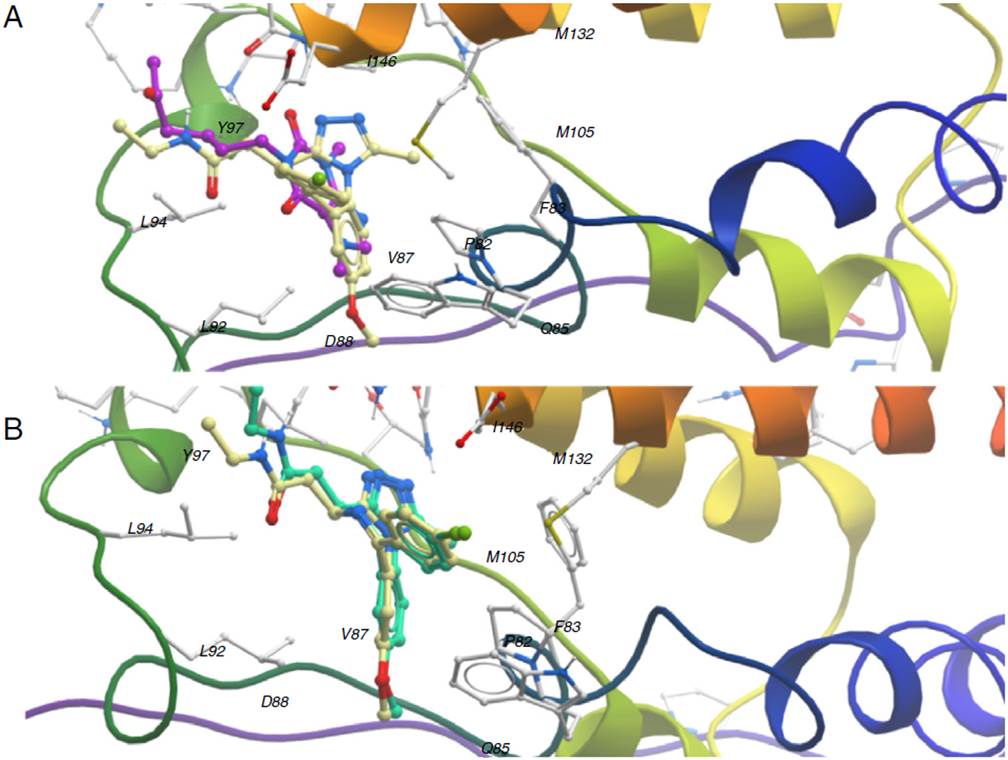
Figura 3 Modelo de acoplamiento de la diana epigenética BRD4 (PDB 3P5O); A) Modelo de acoplamiento de pentoxifilina (átomos de carbono en púrpura) respecto al ligando cocristalizado (amarillo). B) Validación del método de acoplamiento: la conformación en azul es la predicción del algoritmo de docking.
Es posible hacer acoplamiento molecular automatizado entre 2 macromoléculas, como 2 proteínas. Considerando la flexibilidad de las moléculas, el número de posibles conformaciones puede ser muy elevado. De igual manera, si la cavidad del sitio receptor es grande y/o flexible, es más complicado encontrar la posición y orientación que tendrá la molécula pequeña dentro del receptor. Es así que el uso de computadoras ayuda a acelerar el proceso de búsqueda y sugiere modelos de unión.
El acoplamiento molecular automatizado tiene 2 componentes: a) docking o proceso de búsqueda de la conformación y orientación de las moléculas, y b) scoring, que consiste en asignar un valor o puntaje que mida la interacción entre las 2 estructuras.
De las 2 etapas, la segunda es la más difícil de calcular con precisión de forma rápida. Es por esto que en la actualidad el acoplamiento molecular es especialmente útil para proponer modelos de unión, pero aún no son muy confiables para calcular con precisión la energía de interacción. Esto se debe a la gran cantidad de aproximaciones que se hacen para calcular la energía de interacción de forma rápida. Por ejemplo, no se considera en detalle la flexibilidad y solvatación de la diana molecular. Para solucionar este problema los modelos obtenidos por acoplamiento molecular se someten a otros cálculos más refinados que se calculan con niveles de teoría elevados.
Modelado del farmacóforo
Otro método computacional muy utilizado es el modelo del «farmacóforo». Este se define como un arreglo tridimensional de las características mínimas necesarias estéricas y electrónicas para asegurar interacciones óptimas con un blanco farmacológico específico, lo cual desencadenará o bloqueará una respuesta biológica. Aunque la representación visual de un farmacóforo puede ser bi o tridimensional, es importante entender que un farmacóforo no representa a una molécula real o una asociación real de grupos funcionales. Un modelo de farmacóforo es un concepto abstracto que indica la capacidad de interacción molecular común de un grupo de compuestos dirigidos a un blanco farmacológico específico. En otras palabras, el farmacóforo puede ser considerado como el común denominador de un conjunto de moléculas activas.
Cribado virtual
El cribado virtual es un filtrado computacional (in silico) de moléculas para seleccionar candidatos i.e., hits computacionales, para su evaluación experimental (Fig. 4) (Scior et al., 2012). De esta manera, el cribado virtual reduce significativamente el número de ensayos biológicos que se harían si no hubiera una selección de compuestos. Sin embargo, es un proceso predictivo que debe integrarse con ensayos experimentales que validen las predicciones de los ensayos in silico.
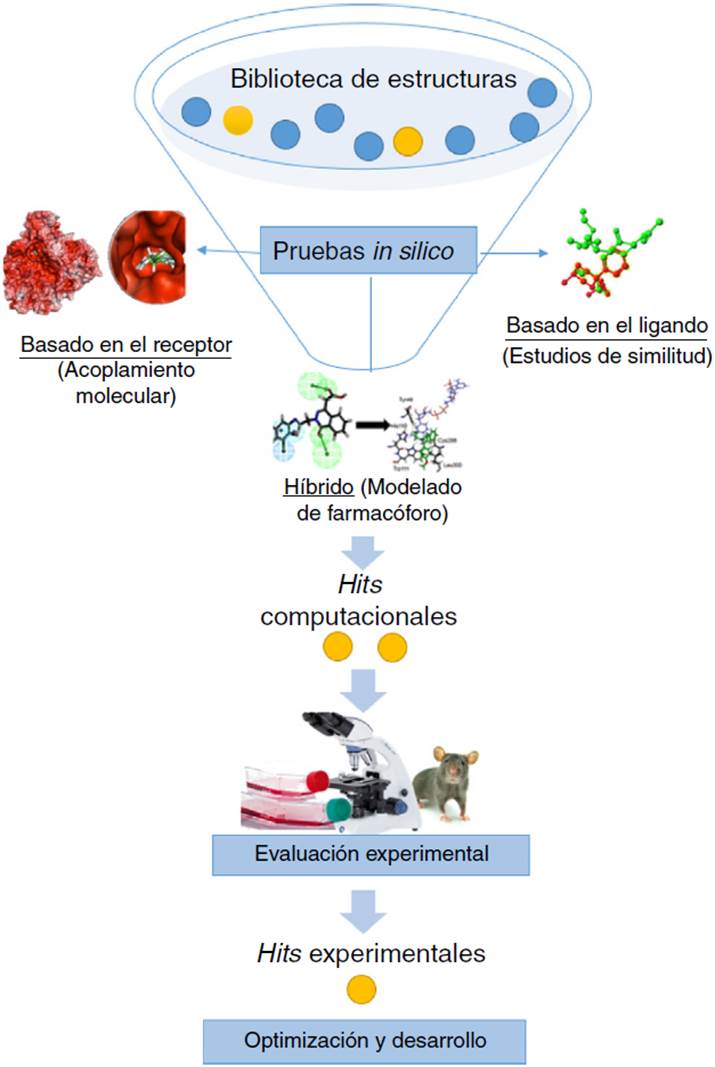
Existen diversos filtros que se utilizan para llevar a cabo el cribado virtual los cuales pueden variar según la complejidad de la base de datos y la información experimental de la que se disponga. Por ejemplo, si se conoce la estructura tridimensional (3D) del receptor se sugiere un cribado basado en la estructura (acoplamiento molecular). Si solo se conocen los compuestos activos, pero no el receptor, entonces la búsqueda se hace basada en el ligando (similitud molecular). Si se conoce la estructura 3D del receptor y de los compuestos activos se pueden combinar filtros para facilitar la búsqueda, tales como descriptores moleculares y propiedades farmacocinéticas, entre otras. Las técnicas para hacer cribado virtual dependen de la información disponible del sistema.
En nuestro grupo de investigación se ha utilizado el cribado virtual usando acoplamiento molecular para la identificación de nuevos inhibidores de las enzimas AKT-2 y ADN metiltransferasa (Kuck, Singh, Lyko y Medina-Franco, 2010; Medina-Franco et al., 2009). Ambas son dianas moleculares para el desarrollo de compuestos contra el cáncer. Los hits experimentales han servido como puntos de partida de programas de optimización (Hernández-Campos et al., 2010; Medina-Franco, Fernández-de-Gortari y Naveja, 2015).
Cribado virtual de fármacos aprobados: reposicionamiento de fármacos
El reposicionamiento de fármacos consiste en encontrar una nueva aplicación terapéutica distinta por la que fue diseñado un fármaco y que no necesariamente tiene que ser la misma dosis. El objetivo de esta estrategia es reducir los gastos y tiempos de investigación. Casos exitosos de reposicionamiento de fármacos se han discutido recientemente (Naveja, Dueñas-González y Medina-Franco, 2016).
A partir de la hipótesis de que «moléculas similares tienen propiedades similares», se han aplicado estrategias computacionales encaminadas a reposicionar fármacos. Un ejemplo es el esfuerzo de reposicionar la olsalazina como anticancerígeno. La olsalazina es un antiinflamatorio aprobado para su uso clínico. La olsalazina fue identificada como un agente hipometilante gracias a la búsqueda de similitud asistida por computadora en DrugBank (Méndez-Lucio, Tran, Medina-Franco, Meurice y Muller, 2014).
Quimioinformática
La quimioinformática es una herramienta que surge de la combinación de recursos informáticos y datos químicos, y se emplea en el manejo, visualización y análisis sistemático de información química. Esta herramienta permite analizar eficientemente miles de datos. A través de métodos quimioinformáticos es posible manejar y organizar información, visualizar el espacio químico, hacer minería de datos y establecer asociaciones matemáticas entre la estructura y actividad. Un ejemplo clásico son las relaciones cuantitativas estructura-actividad (QSAR, por sus siglas en inglés).
Bases de datos públicas
La quimioinformática tiene aplicaciones especialmente valiosas en el análisis y manejo de bases de datos moleculares. Ejemplos de colecciones usadas con frecuencia en descubrimiento de fármacos son ZINC, DrugBank, ChEMBL, Binding Database y PubChem. Estas colecciones discuti-das en otros artículos (Nicola, Liu y Gilson, 2012; Scior, Bernard, Medina-Franco y Maggiora, 2007) contienen información para realizar cribado virtual, quimiogenómica, análisis estructura-actividad y reposicionamientos de fármacos, entre otros.
Análisis computacionales en línea
La información contenida en bases de datos públicas se puede analizar con recursos integrados en la misma base de datos. Un ejemplo de este tipo de plataformas es PubChem BioAssay Database (http://pubchem.ncbi.nlm.nih.gov). PubChem también tiene herramientas que permiten hacer el análisis de todos los datos, tales como análisis de relaciones estructura-actividad y agrupamiento.
Relaciones estructura-actividad
Cuando no se conoce la estructura del receptor se utilizan métodos basados en el ligando, los cuales dependen de la información disponible para estructuras químicas con actividad biológica conocida. Métodos comunes son estudios SAR (cualitativos) y QSAR (cuantitativos).
QSAR y métodos comunes
De acuerdo con el origen de los descriptores moleculares usados en los cálculos, los métodos QSAR pueden dividirse en 3 grupos. Uno de ellos usa un número relativamente pequeño de descriptores que describen, por ejemplo, efectos hidrófobos, estéricos y electrostáticos. Otro grupo se basa en propiedades calculadas a partir de la conectividad molecular. Debido a que las fórmulas estructurales son bidimensionales, estos métodos se conocen como estudios QSAR en 2 dimensiones o QSAR-2D. Un tercer grupo de métodos se basa en los descriptores obtenidos de la representación tridimensional de las estructuras y se les conoce como QSAR en 3 dimensiones o QSAR-3D. Uno de los ejemplos más conocidos de QSAR-3D es el análisis comparativo de campos moleculares (del inglés, comparative molecular field analysis [CoMFA]). Los estudios QSAR también pueden clasificarse por el tipo de métodos de correlación empleados; estos pueden ser métodos lineales, como es el caso de la regresión lineal o regresión lineal múltiple, y regresión no lineal.
Panoramas de actividad
El modelado de panoramas de actividad (en inglés, activity landscape modeling) tiene como objetivo describir sistemáticamente cambios en la actividad biológica asociados con cambios en las estructuras químicas. Una de las aplicaciones es identificar casos en donde pequeños cambios estructurales están asociados a un cambio grande en la actividad biológica. Estos casos llamados «acantilados de actividad» (en inglés, activity cliffs) son excepciones al «principio de similitud» y tienen un impacto importante en el diseño de fármacos (Medina-Franco et al., 2013).
Casos exitosos
Los cálculos computacionales han tenido un papel significativo en la investigación de moléculas que actualmente se encuentran en uso clínico. Por ejemplo, el DIFAC ya ha tenido contribuciones notables en el tratamiento del síndrome de la inmunodeficiencia adquirida, en infecciones por el virus de la influenza y en el tratamiento del glaucoma (Medina-Franco, 2007; Talele, Khedkar y Rigby, 2010). En la Tabla 2 se resumen ejemplos de casos de éxito recientes.
Tabla 2 Ejemplos recientes de fármacos desarrollados con la ayuda de métodos computacionales
| Fármaco | Aplicación | Técnica computacional | Farmacéutica |
|---|---|---|---|
| Crizotinib | Agente antineoplásico (cáncer de pulmón) | Acoplamiento molecular | Pfizer |
| Rilpivirina | Antiviral (infección por el VIH) | Acoplamiento molecular | Janssen Pharmaceuticals |
| Zelboraf | Agente antineoplásico (melanoma) | Acoplamiento molecular y análisis de subestructuras |
Roche |
| Boceprevir | Antiviral (infección por el virus de la hepatitis C, VHC) |
Acoplamiento molecular | Merck & Co |
En agosto de 2011, crizotinib (Xalkori; Pfizer) fue aprobado por la FDA para el tratamiento de pacientes con cáncer de pulmón de células no pequeñas (NSCLC). Crizotinib fue descubierto a través de estudios para identificar inhibidores similares a fármacos potentes de la tirosina quinasa del receptor de MET. Posteriormente, también se observó que era un inhibidor potente de ALK. Este descubrimiento se dio a partir de ensayos biológicos y de estudios de acoplamiento molecular. Actualmente se ha observado un desarrollo de resistencia adquirida hacia el crizotinib, y entre los mecanismos identificados se encuentra la adquisición de mutaciones de resistencia secundaria dentro del dominio de tirosina quinasa de ALK, los cuales también pueden ser estudiados por medio de estudios de acoplamiento molecular y dinámica molecular (Cui et al., 2011).
Conclusiones
En los últimos años el DIFAC ha tenido un desarrollo sin precedentes gracias a los avances de diversas disciplinas como la biología molecular, química teórica, quimioinformática, ciencias de la computación y avances tecnológicos. La incorporación de métodos de cómputo al esfuerzo multidisciplinario que implica el desarrollo de medicamentos ha llevado al desarrollo de fármacos que se encuentran en uso clínico. Además, los métodos de cómputo hacen constantes aportaciones a proyectos de investigación para analizar datos en forma eficiente y plantear hipótesis valiosas que guían el diseño de nuevos fármacos. Se espera que aumente el número y calidad de beneficios que aportan los métodos in silico al diseño de medicamentos.
Perspectivas
El DIFAC tiene aún muchos retos que, sin estar limitado a ellos, incluyen: a) incrementar la eficiencia del cribado virtual; b) aumentar el número y la calidad de los recursos computacionales en línea; c) desarrollar el campo de la quimiogenómica computacional; d) fortalecer el diseno˜ de fármacos dirigidos a múltiples dianas moleculares; e) mejorar la capacidad predictiva de modelos de toxicidad y efectos secundarios, y f) fortalecer la interacción con otras disciplinas para optimizar la búsqueda de moléculas bioactivas para el tratamiento y/o prevención de enfermedades.

