










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de neurociencias (México, D.F.)
versión On-line ISSN 1028-5938versión impresa ISSN 0187-4705
Arch. Neurocien. (Mex., D.F.) vol.10 no.3 Ciudad de México jul. 2005
Artículo de revisión
Cerebral ischemia: some secondary alterations and animal models
Isquemia cerebral: algunas alteraciones secundarias y modelos animales
María Estela López–Hernández, Hugo Solís
Laboratorio de Neurofisiología. Departamento de Anatomía. Facultad de Medicina UNAM.
Correspondencia:
María Estela López–Hernández.
Laboratorio de Neurofisiología. Facultad de Medicina UNAM.
Av. Universidad 3000, Edif. B 4º. Col. Copilco Universidad
04510 México, D.F.
E–mail::estelalopez@correo.unam.mx
Recibido: 18 noviembre 2004.
Aceptado: 31 enero 2005.
RESUMEN
El ataque isquémico cerebral es una de las principales causas de morbilidad y mortalidad en los países industrializados, y genera una enorme carga económica en las familias de los enfermos y en la sociedad. El propósito de esta comunicación es revisar los diferentes modelos animales utilizados para estudiar algunas de las lesiones encefálicas causadas por la isquemia cerebral, y hacer énfasis en la importancia que tiene toda esta información que contribuye a entender los mecanismos de lesión neuronal. Algunos de estos procesos involucrados en el daño neuronal aún no están bien entendidos en la actualidad son motivo de detalladas investigaciones con el objetivo de obtener conocimiento más específico. De esta manera se tendrá una mejor herramienta para prevenir y predecir el valor y el efecto terapéutico de las estrategias utilizadas para el tratamiento en el ser humano.
Palabras clave: flujo sanguíneo cerebral, modelos animales, ataque isquémico, infarto.
ABSTRACT
Stroke is a leading cause of morbidity and mortality in industrialized countries. It imposes an enormous economic burden on the families of patients and on society. The purpose of this review is to give an overview about diverse animal models used to study some brain injury by cerebral ischemia with the aim to stress that this information has contributed to understand the mechanism of the neuronal lesion. Some of these injury processes are not yet completely understood and are now the subject of a more detailed investigation in order to obtain more specific knowledge to prevent and to predict the value and effect of therapeutic approaches inhuman subjects.
Key words: cerebral blood flow, animal models, stroke, ischemic penumbra.
The metabolism of the brain depends exclusively on oxygen and glucose 1–5. Commonly the cerebral blood flow (CBF) is carefully auto–regulated so that the normal human brain receives approximately 55 mL/100 g/min (this is approximately 100 mL/100 g/min in rats). This ensures continuous delivery of oxygen and glucose to energize the brain so that it can maintain cell membrane potentials, synthesize, pack and release neurotransmitters, and support cellular architecture 6, 7. Ischemia is defined as a state of insufficient blood flow 1. Interruption of CBF results in loss of consciousness within the first 10 seconds and cessation of spontaneous and evoked electrical activity within the next 10 seconds3. In ischemia CBF falls and, at approximately30 to 35 mL/l 00 g/min, there is an increase in the extracellular ion concentration (H+). When CBF falls to around 20 mL/100 g/min, intracellular migration of water from extracellular fluid leads to swelling of astrocytes, evoked potentials cannot be elicited and the electroencephalogram (EEG) recording becomes isoelectric. Failure of electrical activity is probably associated with the cessation of orderly synthesis and the release of neurotransmitters. Electrical silence, however, is not synonymous with loss of viability. If a net increase in cerebral water content occurs, this constitutes cytotoxic edema, initially distributed in ischemic areas of gray matter. This may reduce regional blood flow by raising local tissue pressure or by distorting blood vessels and increasing resistance to flow. All flows below 15 mL/1 00 g/min, ionic fluxes increase, extracellular potassium (K+) rises extracellular calcium (Ca2T) falls, and the cell membrane depolarizes 1,6,7,9,10. Complete arrest of the cerebral circulation leads within seconds to cessation of neuronal electrical activity and within a few minutes to deterioration of the energy state and ion homeostasis 11. Increases in intracellular Ca2+ is a pivotal event in cellular dysfunction during hypoxia. Cellular dysfunction occurs when anaerobic sources of energy fail to maintain energy production at the level required by the various metabolic processes. Irreversible cellular damage occurs when the depletion of energy affects the reactions responsible for maintaining cellular integrity. Such is the inevitable sequence of events when the blood flow to the brain is arrested2,3,6,7,9(figure 1).
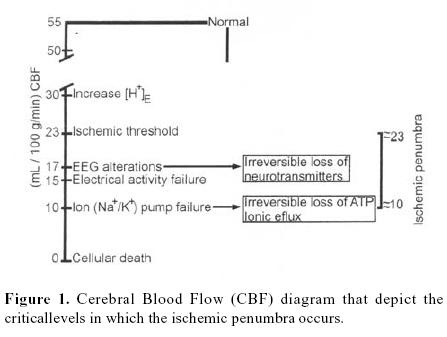
Some evidence indicates that in immediate failure of basic functions such as synaptic transmission, ion pumping and energy metabolism in the ischemic brain is critically dependent on residual blood flow, and that these functions tail at certain critical flow thresholds. The condition of the ischemic brain with flow between the two thresholds –the upper threshold of electrical failure and the lower threshold of energy failure and ion pump failure– can be described by electrical silence with normal or only slightly elevated extracellular K+ concentration. The residual perfusión supplies sufficient oxygen to maintain a close to normal tissue concentration of adenosin triphosphate (ATP). Since the concentrations of phosphorcreatine and lactate are greatly reduced and increased, respectively, and since the concentrations of adenosin dephosphate (ADP) and adenosin monophosphate (AMP) are moderately increased, some degree of energy failure exists. It appears, further, that the development of infarction is critically correlated to residual perfusión, and there is a lethal threshold of residual blood flow below which tissue infarction develops after a certain time 11. Results obtained in hypoxia suggest that such moderate energy imbalance does no lead to neuronal damage. In focal ischemia the tissue in this condition forms a ring around the more densely ischemic center, in which energy failure and ion pump failure have developed. In analogy to the half–shaded zone around the center of a complete solar eclipse this part of the ischemic brain has been termed as the <<penumbra>>. This term is descriptive only, and may equally well be applied in global ischemia. Although rather labile in the epileptic rat brain, the state of the <<penumbra>> seems stable for hours in focal ischemia and its identification may be valuable in experimental and even clinical conditions 5,9,11,13. Brain ischemia can result from a wide range of disturbances including cardiovascular and respiratory disorders, brain trauma14, and any condition leading to prolonged arterial hypotension or intracranial hypertension. Even in the absence of disease, cerebral ischemia remains a potential hazard of general anaesthesia79.
Classification of cerebral ischemia:
The magnitude of ischemic cerebral damage relates the duration to the severity of ischemia. One way to classify this alteration is: Primary brain ischemia, that occurs because of regional cerebral ischemia (embolic or focal stroke) or global hypoxia/ischemia (resuscitation from cardiac arrest, near drowning, carbon monoxide poisoning or massive hemorrhage). Secondary brain ichemia, occurs in tandem with the brain injury caused by trauma, tumor or infection or with vasospasm following subarachnoid hemorrhage and is often the mayor cause of morbidity and mortality in these conditions7,9. The pattern of ischemic cerebral damage depends on whether ischemic is global or focal complete or incomplete. The ischemic insults and animal models are traditionally divided into two types: global and focal (table 1). Global ischaemia may be incomplete –for example, in oligoemic hypoxia arising during induced or posttraumatic arterial hypotension, or in carbon monoxide poisoning (an instance of anaemic hypoxia)– or complete after cardiac arrest (stagnant anoxia). Incomplete global ischemia implies maintenance of some flow, and ischemic damage is typically distributed along the boundary zones of flow between the main cerebral arteries. Complete global anoxia induces damage predominantly in the most recently evolved areas of the brain–neocortex, basal ganglia, hippocampus and cerebellum. While complete functional recovery cannot be expected after more than 7 minutes of normothermic global anoxia some neurons survive 1 hour of circulatory arrest6,9,15. As a general rule, under normothermic conditions, 10 min. of global ischemia are lethal in man5,9. Focal ischemia that follows transient or permanent flow reduction in the territory of a cerebral artery is invariably incomplete, and arises when blood flow in a supplying artery is reduced or stopped by atheroma, embolism, inflammatory disease or trauma (oligaemic hypoxia). The amount of damage depends on the state of the systemic or collateral circulation, and will be increased by seizure activity, hypoxaemia and systemic hypotension. Focal ischemia may also develop during episodes of raised intracranial pressure after severe head injury and it is characterized by the formation of an ischemic penumbra5,7,9,11,15. If the ischemia is incomplete the outcome is more difficult to predict and is largely dependent on residual perfusión and oxygen availability. It is in large measure the outcome of incomplete cerebral ischemia, which is of particular interest in cerebral vascular disease5,7,9,11,15.
Animal Models
Ischemic stroke is one of the major causes of long–term neurological disability. This condition can be reproduced in rodents and larger animals using well–established procedures, many of which were originally developed in rats16,18. The use of animal model systems forms a fundamental part of neuroscience research efforts to improve the prevention, diagnosis, understanding and treatment of neurological conditions. Without such models it would be impossible to investigate such topics as the underlying mechanisms of neuronal cell damage and death orto screen compounds for possible anticonvulsant propellies19-22. Many of the experimental approaches have been provoked by the results of investigations that used dissociated cell culture, neuronal cell lines or hyppocampal slices to study hypoxia or hypoxia/ aglycemia in vitro, but these <<test tube>> lacks many features relevant to cerebral ischemia in vivo. In vivo methods that have been adopted for modeling stroke have been subject to much criticism, but consistency can be achieved, particularly in rat models, with minimal variation from laboratory to laboratory. Rodent models can be used to mimic changes in regional CBF and cerebral metabolism in a way that simulates what we have learned from human stroke by means of positron emission tomography and magnetic resonance spectroscopy. Similarity in the appearance, topography and temporal evolution of the radiologic and histopathologic injury lends further credence to the appropriateness of rodent models and suggests that agents which prevent brain injury in rats are worth testing in humans5,7.
Although stroke has been studied in many species (gerbil, cats, rabbits, dogs, baboons) rats and mice are the most widely investigated 20,23. Rats are the most commonly used species for the modeling of neurological disease. In addition to providing an indispensable tool for basic research, rat models of human disorders allow us to investigate therapeutic strategies as a prerequisite to their testing in patients 5,18,20. Mice are specially useful because of the availability of unique strains that can be generically engineered to over or under express targeted gene5,24,27.
Several well established models are available to study ischemia. In relation to the etiology or mechanism of ischemia two major approaches are available for disruption of the circulation of healthy blood to the brain: extravascular arterial compression by extrinsic ligation, and manipulation of the intravascular hemodynamics by embolism or hypotension. Extravascular occlusions occur in humans during surgical treatment; in experimental animals extravascular compression produces acute ischemia in the vascular distribution distal to the clip or ligature. Small spring clips may be applied under direct visual control so that the precise site of vascular occlusion is known. Furthermore, clips may be removed after known time intervals and therefore may be used in modeling temporary or transient ischemia. However, intracranial arterial occlusion requires craniectomy, and the direct disruption of the autonomic nerve supply to the vascular bed seems unavoidable and may introduce physiological and biochemical artifacts. Intravascular methods spare the arterial nerve supply, do not require craniectomy, and while usually irreversible, may be used for pathophysiological studies spanning acute and chronic phases. Nonetheless, the site of vascular occlusion is controlled only by the size and physical characteristics of the embolic substance used. Embolic material must pass through a rete mirabile in all laboratory species other than dogs and primates in order to occlude intracranial vessels 5,28. In vivo global models are created by cardiac arrests with resuscitation 29,34 and by transiently occluding both common carotid arteries (CCA) in association with either hypotension or prior bilateral vertebral artery occlusion. In the so called four vessel occlusion model, flow in both CCA and vertebral arteries are blocked for a specified time period35–37. Three vessel occlusion model may be performed by ligation of one middle cerebral artery (MCA) and both CCA in succession or by occlusion of the basilar artery with a miniature clip followed by bilateral CCA27,38. In the two vessel occlusion model, which is also referred as severe forebrain ischemia, only the CCA are temporarily occluded39–41, sometimes along with mild hypotension42. In all of these models injury develops selectively in cells most vulnerable to ischemic damage such as in the CAI sector in hippocampus, medium sized neurons in the striatum, and Purkinje cells in cerebellum. The variations differ relation to time and type of ischemia41 in CBF, neuronal excitability, in number, and in qualitative changes in the neurons of hippocampus differ. Experimental focal ischemia is most commonly studied during permanent or transient occlusion of the MCA43–44. Proximal MCA occlusion can be induced by an intraluminal suture (so–called, filament model)45–47or with a vascular clip and it causes injury to cortex and deep structures (striatum). Distal MCA occlusion (the so–called Brint model) is usually produced by placing a vascular clip on a pial vessel or by cautery48. The occlusion typically spares stratium and primarily involves the neocortex. Pannecrosis develops in the territory supplied by the respective artery with glial and endothelial cell death. If recirculation is established early (2 hours or less) the outcome is better (transient MCA occlusion)49. In some ways, the reperfused brain imitates restoration of blood flow after spontaneous lysis of a thrombo–embolic clot in humans, even though reperfusion after clot lysis is certainly more complex than an on/off phenomenon as modeled by placement and retraction of an intravascular filament5,28,45,47. In vitro models allow to study the effects of cerebral ischemia in post mitotic neurons and the so–called oxygen glucose deprivation (OGD) is the model commonly used. Primary cultures of post mitotic neurons from different regions of the brain (such as cortex, striatum, septum, hippocampus, etc) can be established from rat or mouse embryos (day 16 to 18). After several days in vitro (1st to 14th day) these post mitotic cells can be exposed to a combined deprivation of oxygen and glucose. Depending on the length and severity of the insult, cell death develops and can be quantified on a morphological, biochemical or molecular basis50–51. Other possibilities to study ischemic damage in vitro include the studies of brain slices, particularly the hippocampal slice52–55. Usually, the bathing solution is changed from a mixture of oxygen/carbon dioxide to nitrogen/carbon dioxide in the absence (hence ischemia) or presence (hence anoxia) of glucose. Generally, 5–7 min of ischemia can lead to profound cell loss in the CA1 region. Shorter insults lead to a more slowly evolving damage, requiring approximately 12 h to be manifested. This type of cell death, resembles in many ways, the delayed neuronal death seen in vivo5.
Secondary alterations by cerebral ischemia
The typical histological picture following global ischemic insults is described by delayed neuronal death sparing glial cells (sometimes even associated with astrogliosis)5. Factors responsible for cell death in brain ischemia are believed to involve aberrations in cellular metabolic pathways, specifically calcium, arachidonic acid, oxygen metabolism and acid–base regulation15. Global ischemia is followed by selective ischemic necrosis of neurons (SINN). SINN is profound for certain populations of neurons, such as the hippocampus CA1 pyramidal cells, cortical neurons in layers 3, 5, and 6, cerebellar Purkinje cells, and the small and medium sized striatal neurons36. MCA occlusion produces a relative reduction of regional CBF, which has to be maintained for at least 1 to 2 hours followed by a period of reperfusion (temporary focal ischemia) or for 3 to 6 hours or longer (permanent focal ischemia)7. If ischemia is prolonged, the ischemic brain is rendered inordinately acidotic or hyperthermic and the failure of adequate reperfusion injury is more likely to result in infarction, defined as a volume of brain tissue in which there is pannecrosis, i.e. the death of neurons, glia and endothelial cells56-60. The process of SINN differs from infarction not only because it results from a very brief (5–15 min) but severe global ischemic insult as opposed to a more prolonged, regional and less severe ischemic insult, but also because it requires a prolonged interval of reperfusion (1–7 days) for the full maturation of injury to occur, particularly for cells in the hippocampus. The final common pathway appears to be mediated by an lethal intracellular Ca2+ current. It might be conjectured that ischemic brain injury is a continuum with SINN at one extreme, with slow degeneration of individual strokesensitive neurons in particular regions, and liquefying pannecrosis at the other extreme. An explanation for the conundrum of SINN would help rationalize attempts to develop an effective pharmacotherapy for acute stroke7,36,38,57,59,60. Transient forebrain ischemia results in an initial energy failure, followed by a homogeneous restoration of blood flow which results in recovery of energy charge, electrophysiologic function, and membrane and neurotransmitter function, but subsequently leads to the death of certain cells in a delayed fashion7. After longer times of ischemia, reperfusion is incomplete due to microvascular occlusion which was termed as the no–reflow phenomenon some thrirty years ago61. Following significant ischemic insults, progressive brain hypoperfusion ensues, beginning 15–120 min after reperfusion. Hypoperfusion results from elated cerebral vascular resistance, as has been shown to occur at the precapillary and capillary level15,45–47,62,63. Following ischemia, endothelin may be produced by damaged indothelial cells, causing powerful vasoconstriction, and this may be mediated, in part, by an endothelium derived platelet activating factor (PAF), which can also be derived from platelets7,61. Furthermore, although increased blood viscosity may contribute to impaired microvascular perfusión, clotting does not appear contributory and heparin has no beneficial effect15,61,64. Ischemic insults lead to a heterogeneous distribution of injury15. Selective vulnerability to cerebral hypoxia exists not only between different cellular elements in the brain, neurons being most sensitive and microglia and blood vessel cells least sensitive, but also between different neurons9. Neurons are more susceptible than glial cells, and die over hours to days after the insult5. Selective vulnerability most likely results primarily from differences at the cellular level. Secondary vascular compromise probably occurs during the reperfusion period, presumably from cellular changes associated with ischemia and reperfusion15. In gerbils subjected to 5 mil of forebrain ischemia CA 1 neurons disintegrate after a delay of 2–3 days (delayed neuronal death)65,66. Similarly, in rats submitted to forebrain ischemia of 10–30 min, neuronal death appears after delayed of 1–3 days36,67–70.
Regional idiosyncrasies of postischemic CBF and metabolism are two factors that might explain the greater sensitivity of certain neurons to ischemic damage and also the progressive nature of neuronal damage in some brain regions37.
Cellular damage during reperfusion following either global or focal ischemia may be influenced by substances that accumulate during the early reperfusion period. During reperfusion, free radical production and nitric oxide (NO) generation are especially pronounced and contribute to reperfusion injury5,7,62,63,71,72. The production of oxy radicals and the products of lipid peroxidation suggests a role for lipid peroxidation inhibitors as a therapy for reducing brain injury following ischemia. Lipid peroxidaton products are increased following transient ischemia but remain unchanged in regions of the brain exposed to permanent focal occlusion suggesting that lipid peroxidation inhibitors are more efficacious following transient rather than permanent ischemia7,63.
The hippocampus exhibits the highest sensitivity of ischemia throughout the central nervous system, and within the hippocampus the hilus and the CAI sector are much more sensitive than the CA3 sector or the dentate gyrus66. Delayed neuronal death in hippocampus has also been observed in patients who survived cardiac arrest for more than 24 h73,74. Several molecular factors have been proposed to contribute to hippocampal injury75, such as glutamate mediated excitation76–80, calcium toxity81,82 down–regulation of adenosin receptors83, inhibition of protein synthesis84,85, or disturbances of polyamine metabolism86. However, hemodynamic mechanisms cannot be excluded87,88, and it has not been established which of these factors is the limiting pathophysiological event and how they relate to the duration of ischemia. In fact, selective injury of the different sectors of hippocampus is usually documented after relatively short periods of cerebral ischemia or with an interval (7 days) of reperfusion41,67,89– 93, whereas other neuronal populations are only involved when ischemia is prolonged or complicated by hyperglycemia3,70,93. Because oxygen free radicals and NO promote apoptotic cell death94–96, transient ischemia models have become especially useful to investigate cell death in vivo97,98 which may particularly apply for models of mild ischemia99–102. In these models apoptosis is prominent after 30 min MCA occlusion followed by longer reperfusion times (several days)102,103. The pattern of cell death is reminiscent of global ischemia in that it is both selective for neurons and delayed. Mild ischemia models may be similar to transient ischemic attacks in man. In fact, changes in T1/T2 weighted magnetic resonance imaging 7 days after 15min MCA occlusion in rats resemble those 7 to 10 days after transient ischemic attacks TIAs in patients with known cardiogenic embolism. However, selective neuronal death has not yet been convincingly documented following TIAs in humans5.
CONCLUSIONS
Global and focal cerebral ischemia represents diseases that are common in the human population. Animal models have contributed to our understanding about the diverse factors of neuronal cell damage and death for cerebral ischemia in the human. We have come to infer that not only the type and duration of ischemia is the event of major significance, but also that once reperfused, reoxygenation causes further injury. Free radical generation and other biochemical compounds following reperfusion liberate different metabolic processes and contribute on diverse ways to cell membrane injury that we can see on dissimilar manners like electrophysiological failure. These observations have led to rational theoretic approaches for the prevention, diagnosis and treatment of cerebral ischemic brain injury.
REFERENCES
1. Hansen AJ. Effect of anoxia on Ion distribution in the brain. Physiol Rev 1985; 65(1):101–48. [ Links ]
2. Kass IS, Abramowicz AE, Cottrell JE. Brain metabolism during ischemia and anoxia. J Neurosurg Anest 1989; 1 (3) :264–66. [ Links ]
3. Gutierrez G. "Cellular energy metabolism during hypoxia. Crit Care Med 1991; 19 (5):619–26. [ Links ]
4. López–Hernández E, Solís H. Generalidades sobre el metabolismo cerebral relacionadas con la isquemia–anoxia. Rev Mex Enf Cardiol 1995;3(4):93–7. [ Links ]
5. Endres M, Dimagl U. Ischemia and Stroke. Adv Exp Med Biol 2002; 513:455–73. [ Links ]
6. Navarro RCE. Metabolismo cerebral en isquemia. En: Velez AH, Rojas MW, Borrero RJY, Restrepo MJ. Eds. Fundamentos de medicina. Neurología. Corporación para Investigaciones Biológicas. Medellín, Colombia 1991. [ Links ]
7. Buchan A. Advances in cerebral ischemia: experimental Approaches. Neurol Clinics 1992; 10(1):49–61. [ Links ]
8. Raichle ME. The pathophysiology of brain ischemia. Ann Neurol 1983; 13:2–10. [ Links ]
9. Dearden NM. Ischaemic brain. Lancet 1985; 3:255–59. [ Links ]
10. Martínez RHR. Fisiopatología de la isquemia cerebral. En: Barinagarrementeria F. (Ed). Enfermedad vascular cerebral. Instituto Syntex. México 1991. [ Links ]
11. Astrup I, Siesjo BK, Symon L. Thresholds in cerebral ischemia. The ischemic penumbra. Stroke 1981 ;12(6):723–25. [ Links ]
12. Fisher M. The ischemic penumbra: identification, evolution and treatment concepts. Cerebrovasc Dis 2004; 17 Suppl 1: 1–6. [ Links ]
13. Lovblad KO, Koussy ME, Oswald H, Baird AE, Schroth G. Magnetic resonance imaging oft he ischaemic penumbra. Swiss Med Wkly 2003; 133:551 –59. [ Links ]
14. BramlettHM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood FlowMetab 2004; 24(2): 133–50. [ Links ]
15. Kaplan J, Dimlich RVW, Biros MH, Hedges J. Mechanisms of ischemic cerebral injury. Resuscit 1987;15:149–69. [ Links ]
16. Alonso de Lecinana M, Diez–Tejedor E, Carceller F, Roda JM. Cerebral ischemia: from animal 13 studies to clinical practice. Should the methods be reviewed? Cerebrovasc Dis 2001; 11 Suppl :20–30. [ Links ]
17. Fuxe K, Bjelke B, Andbjer B, GralmH, Rimondini R. Endothelin–1 induced lesions of the frontoparietal cortex of the rat. A possible model of focal cortical ischemia. Neurorep 1997; 8:2623–9. [ Links ]
18. Cenci MA, Whishaw IQ, Schallert T. Animal models of neurological deficits: how relevant is the rat? Nature 2002; 3:574–9. [ Links ]
19. Lythgoe MF, Sibson NR, Harris NG. Neuroimaging of animal models of brain disease. Br Med Bull 2003;65:235–57. [ Links ]
20. Roda JM, Carceller F, Pascual JM, Herguido MJ, González–Llanos F. Modelos animales experimentales en isquemia cerebral. Neurología 1998;13(9):427–30. [ Links ]
21.Traystman RJ. Animal models of focal and global cerebral ischemia. ILARJ 2003;44(2):85–95. [ Links ]
22. Leker RR, Constantini S. Experimental models in focal cerebral ischemia: are we there yet? Acta Neurochri Suppl 2002; 83:55–9. [ Links ]
23. Ginsberg MD, Busto R. Rodent models of cerebral ischemia. Stroke 1989;20(12): 1627–42. [ Links ]
24. Fassler R, Martin K, Forsberg E, Litzenburger T, Iglesias A. Knockout mice: how to make them and why. The immunological approach. Int Arch Allergy Immunol 1995; 106(4):323–34. [ Links ]
25. Majzoub JA, Muglia LJ. Molecular medicine knockout mice. N Engl J Med 1996; 334(14):904–07. [ Links ]
26. Rubin EM, Barsh GS. Perspectives series: molecular medicine in genetically engineered animals. J Clin Invest 1996; 97(2) :275–80. [ Links ]
27. Yonekura I, Kawahara N, Nakatomi H, Furuya J, Kirino T. A Model of global cerebral ischemia in C57 BL/6 mice. J Cereb Blood Flow Metab 2004; 24(2):151–8. [ Links ]
28. Molinari GF. and Laurent JP. A Classification of experimental models of brain ischemia. Stroke 1976; 7(1):14–7. [ Links ]
29. Pluta R, Lossinsky AS, Mossakowski MJ, Faso L, Wisniewski HM. Reassessment of a new model of complete cerebral ischemia in rats. Acta Neuropathol 1991 ;83:1–11. [ Links ]
30. Gisvold SE, Safar P, Hendrickx HHL, Rao G, Moossy J. Thiopental treatment after global brain ischemia in pigtailed monkeys. Anesthesiol 1984;60:88–96. [ Links ]
31. Cervantes M, Ruelas R, Sánchez R, Alvarez–Resendiz G. Brain injury following cardiorespiratory arrest in cats. Effects of alphaxalone–alphadolone. Bol Estud Méd Biol Mex 1989;37:17–27. [ Links ]
32. Cervantes AJM. Análisis comparativo de la actividad eléctrica de diversas estructuras cerebrales en gatos sometidos a isquemia–anoxia cerebral global aguda. Efecto de la nimodipina. Tesis de Doctorado (Área Farmacología). Facultad de Medicina, UNAM 1993. [ Links ]
33. Contreras GA. Análisis electrográfico del efecto de la naloxona sobre varias estructuras cerebrales en gatos sometidos a isquemia–anoxia cerebral global aguda. Tesis de Doctorado en Ciencias Fisiológicas. UACPyP del CCH. Centro de Neurobiología. UN AM 1998. [ Links ]
34. González VMD. Análisis del efecto protector de la progesterona contra el daño provocado por isquemia–anoxia cerebral global aguda en el gato. Tesis de doctorado en ciencias fisiológicas. UACPyP del CCH. Centro de Neurobiología. UNAM 2000. [ Links ]
35. Puisinelli W A, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetizad rat. Stroke 1979; 1 0(3):267–72. [ Links ]
36. Puisinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient fore brain ischemia. Ann Neurol 1982; 11(5):491–8. [ Links ]
37. Puisinelli W A, Levy DE, Duffy TE. Regional cerebral blood flow and glucose metabolism following transient forebrain ischemia. Ann Neurol 1982 ;11 (5) :499–509. [ Links ]
38. Chen ST, Hsu CY, Hogan EL, Maricq H, Balentine JD. A model of focal ischemic stroke in the rat: reproducible extensive cortical infarction. Stroke 1986; 17(4):738–43. [ Links ]
39. Eklöf B, Siesjo BK. The effect of bilateral carotid artery ligation upon the blood flow and the energy state of the rat brain. Acta Physiol Scand 1972; 86: 155–65. [ Links ]
40. Eklöf B, Siesjó BK. The effect of bilateral carotid artery ligation upon acid–base parameters and substrate levels in the rat brain. Acta Physiol Scand 1972; 86:528–38. [ Links ]
41. López–Hernández E. Alteraciones en la inhibición recurrente y en la cito arquitectura del hipocampo en un modelo de isquemia–hipoxia. Tesis de maestría en ciencias fisiológicas. UACPyP del CCH. Centro de Neurobiología. UNAM 1997. [ Links ]
42. Smith ML, Bendek G, Dahlgren N, Rosen I, Wieloch T. Models for studying long term recovery following forebrain ischemia in the rat. 2. A 2–vessel occlusion model. Acta Neurol Scand 1984; 69:385–401. [ Links ]
43.Tamura A, Graham Dl, McCulloch J, Teasdale GM. Focal cerebral ischemia in the rat: description of technique and early neuropathological consequences following middle cerebral artery occlusion. J Cereb Blood Flow Metab 1981 ;1 (1):53–60. [ Links ]
44. Lee EJ, Wu TS, Lee MY, Chen TY, Tsai YY. Delayed treatment with melatonin enhances electophysiological recovery following transient focal cerebral ischemia in rats. J Pineal Res 2004; 36(1):33–42. [ Links ]
45. Zea LE, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989; 20(1 ):84–91. [ Links ]
46. McColl BW, Carswell HV, Mc Culloch J, Horsburgh K. Extension of cerebral hypoperfusion and ischemic pathology beyond MCA territory after intraluminal filament occlusion in C57 BI/6J mice. Brain Res 2004; 997:14–22. [ Links ]
47. Hartings JA, Rolli ML, May Lu XC, Tortella F. Delayed secondary phase of peri–infarct depolarizations after focal cerebral Ischemia: relation to infarct growth and neuroprotection. J Neurosci 2003; 23(37) :11602–10. [ Links ]
48. Brint S, Jacewicz M, Kiessling M, Tanabe J, Puisinelli W. Focal brain ischemia in the rat: methods for reproducible neocortical infarction using tandem occlusion of the distal middle cerebral and ipsilateral common carotid arteries. J Cereb Blood Flow Metab 1988; 8(4):474–85. [ Links ]
49. Hara H, Huang PL, Panahian N, Fishman MC, Moskowitz MA. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J Cereb Blood Flow Metab 1996; 16(4) :605–11. [ Links ]
50. Dawson VL, Kizushi VM, Huang PL, Snyder SH, Dawson TM. Resistance to neurotoxicity in cortical cultures from neuronal nitric–oxide–synthase–deficient mice. J Neurosci 1996; 16(8):2479–87. [ Links ]
51. Rothman S. Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J Neurosci 1984, 4(7): 1884–91. [ Links ]
52.Traynelis SF, Dingledine R. Potassium–Induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol 1988; 59(1 ):259–76. [ Links ]
53. Watson PL, Weiner JL, Carien PL. Effects of variations in hippocampal slice preparation protocol on the electrophysiological stability, epileptogenicity and graded hypoxia responses of CAI neurons. Brain Res 1997; 775:134–43. [ Links ]
54.Tsubokawa H, Oguro K, Masuzawa T, Kawai N. Spontaneous excitatory postsynaptic currents in hippocampal CAI pyramidal neurons of the gerbil after transient ischemia. Neurosci Lett 1995; 191:95–8. [ Links ]
55. Khazipov K, Bregestovski Pand Ben–Ari Y. Hippocampal inhibitory intemeurons are functionally disconnected from excitatory inputs by anoxia. J Neurophysiol 1993; 70(6) :2251 –9. [ Links ]
56. Marcoux FW, Morawetz RB, Crowell RM, DeGirolami U, Halsey JB. Differential regional vulnerability in transient focal cerebral ischemia. Stroke 1982; 13(3): 339–46. [ Links ]
57. Puisinelli WA. Selective neuronal vulnerability: morphological and molecular characteristics. Prog Brain Res 1985; 63:29–37. [ Links ]
58. Torvik A, Svindland A. Is there a transitional zone between brain infarcts and the suitounding brain? A histological study. Acta Neurol Scand 1986; 74(5) :365–70. [ Links ]
59. Nedergaard M. Mechanisms of brain damage in focal cerebral ischemia. Acta Neurol Scand 1988; 77:81 –101. [ Links ]
60. Graham Dl. Hypoxia and vascular disorders. In: Adams JH, Duchen LW (Eds.) Greenfild's neuropathology. Edward Arnold. Great Britain 1992. [ Links ]
61. Ames III A, Wright L, Kowada M, Thurston JM, Majno G. Cerebral ischemia II. The no–reflow phenomenon. Am J Pathol 1968; 52(2):437–53. [ Links ]
62. White BC, Grossman LI, O'Neil BJ, DeGracia DJ, Neumar RW. Global Brain Ischemia and Reperfusion. Ann Emerg Med 1996; 27:588–94. [ Links ]
63. White BC, Grossman LI, Krause GS. Brain injury by global ischemia and reperfusion: A theoretical perspective on membrane damage and repair. Neurology 1993;43:1656–65. [ Links ]
64. Gadzinski DS, White BC, Hoehner PJ, Hoelmer T, Krome C. Canine cerebral cortical blood flow and vascular resistance post cardiac arrest. Ann Emerg Med 1982; 11 (2):58–63. [ Links ]
65. Schmidt–Kastner R, Ophoff BG, Hossmann KA. Pattern of neuronal vulnerability in the cat hippocampus after one hourof global cerebral ischemia. Acta neuropathol 1990; 79:44–55. [ Links ]
66. Kirino T. Delayed neuronal death in the gerbil hippocampus following Ischemia. Brain Res 1982; 239:57–69. [ Links ]
67. Kirino T, Tamura A, Sano K. Delayed neuronal death in the rat hippocampus following transient forebrain ischemia. Acta Neuropathol (Berl) 1984; 64:139–47. [ Links ]
68. Petito CK, Puisinelli W A. Delayed neuronal recovery and neuronal death in rat hippocampus following severe cerebral ischemia: possible relationship to abnormalities in neuronal processes. J Cereb Blood flow Metab 1984; 4(2) :194–205. [ Links ]
69. Sclmlidt–Kastner R, Hossman KA. Distribution of ischemic neuronal damage in the dorsal hippocampus of rat. Acta Neuropathol 1988; 76:411 –21. [ Links ]
70. Smith M–L, Kalimo H, Warner DS, Siesjo BK. Morphological lesions in the brain preceding the development of postischemic seizures. Acta Neuropathol 1988; 76:253–64. [ Links ]
71. Hallenbeck JM, Dutka AJ. Background review and current concepts of reperfusion Injury. Arch Neurol 1990; 47:1245–54. [ Links ]
72.Yulug B, Kilic Ü, Kilic E, Bahr M. Rifampicin attenuates brain damage in focal ischemia. Brain Res 2004; 996:76–80. [ Links ]
73. Fujioka M, Nishio K, Miyamoto S, Hiramatsu K–l, Sakaki T. Hippocampal damage in the human brain after cardiac arrest. Cerebrovasc Dis 2000; 1 0:2–7. [ Links ]
74. Perito CK, Feldmaml E, Puisinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology 1987; 37:1281–86. [ Links ]
75. Abe K, Aoki M, Kawagoe J, Yoshida T, Hattori A Ischemic delayed neuronal death a mitochondrial hypothesis. Stroke 1995; 26(8): 1478–89. [ Links ]
76. Jorgensen MB, Diemer NH. Selective neuron loss after cerebral ischemia in the rat: possible role of transmitter glutamate. Acta Neurol Scand 1982; 66(5):536–46. [ Links ]
77. Simon RP, Swan JH, Griffith T, Meldrum BS. Blockade of N–methyl–D–aspartate receptors may protect against ischemic damage in the brain. Science 1984; 226:850–2. [ Links ]
78. Swan JH, Evans MC, Meldrum BS. Long–term development of selective neuronal loss and the mechanism of protection by 2–amino–7 –phosphonoheptanoate in a rat model of incomplete forebrain ischaemia. J Cereb Blood Flow Metab 1988;8 (1):64–78. [ Links ]
79. Wieloch T. Neurochemical correlates to selective neuronal vulnerability. Prog Brain Res 1985;63:69–85. [ Links ]
80. Wieloeh T, Lindvall O, Blomqvist P, Gage FH. Evidenee for amelioration of isehaemie neuronal damage in the hippoeampal formation by lesions of the perforant path. Neurol Res 1985; 7(1):24–6. [ Links ]
81. Simon RP, Griffith T, Evans MC, Swan JH, Meldrum BS. Calcium overload in selectively vulnerable neurons of the hippocampus during and after ischemia: an electron microscopy study in the rat. J Cereb Blood Flow Metab 1984 ;4(3) :350–61. [ Links ]
82. Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 1995; 268:239–47. [ Links ]
83. Lee KS, Tezlaff W, Kreutzberg GW. Rapid down regulation of hippoeampal adenosine receptors following brief anoxia. Brain Res 1986; 380(1):155–8. [ Links ]
84. Bodsch W, Barbier A, Oehmichen M, Grosse Ophoff B, Hossman KA. Recovery of monkey brain after prolonged ischemia. Protein synthesis and morphological alterations. J Cereb Blood Flow Metab 1986; 6(1):22–33. [ Links ]
85. Thilmann R, Xie Y, Kleihues P, Kiessling M. Persistent inhibition of protein synthesis precedes delayed neuronal death in postischemic gerbil hippocampus. Acta Neuropathol (Berl) 1986; 71 (1–2): 88–93. [ Links ]
86. Pasehen W, Schmidt–Kastner R, Djuricie B, Méese C, Linn F. Polyamine changes in reversible cerebral ischemia. J Neurochem 1987; 49(1):35–37. [ Links ]
87. Imdahl A, Hossman KA. Morphometric evaluation of postischemic capillary perfusión in selectively vulnerable areas of gerbil brain. Acta Neuropathol (Berl) 1986; 69(3–4):267–71. [ Links ]
88. Marinkovic SV, Milisavljevic MM, Vuckovic VD. Microvascular anatomy of the uncus and the parahippocampal gyms. Neurosurgery 1991; 29(6):805–14. [ Links ]
89. Kirino T, Sano K. Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta Neuropathol (Berl) 1984; 62:201–8. [ Links ]
90. López–Hernández E, Parra GL, Bravo J, Reyes TGJ, Solís H. Cambios en la excitabilidad neuronal y alteraciones en la densidad neuronal del hipocampo inducidos por isquemia focal. Arch Neurocien (Mex) 1997; 2(2):3–8. [ Links ]
91. Akai F, Yanagihara T. Identity of the dorsal hippoeampal region most vulnerable to cerebral ischemia. Brain Res 1993; 603:87–95 [ Links ]
92. Solís H, López E, Parra L, Bravo J, Escobar A. Morphological and electrophysiological changes after cerebral ischemia in the rat hippocampus. Exp Neurol 1998; 151(1):170–1. [ Links ]
93. López–Hernández E, Parra GL, Bravo MJ, Garcia HA, Ortiz EA. Repercusiones en el hipocampo de la isquemia cerebral focal transitoria. Rev Mex Enf Cardiol 1997; 5(4):102–8. [ Links ]
94. Naveiro J, Castillo J, Suárez P, Aldrey JM, Lema M Tiempo para el daño cerebral por hiperglicemia en la isquemia aguda. Rev Neurol 1998; 26(153):790–93. [ Links ]
95. Kuschinsky W, Gillardon F. Apoptosis and cerebral ischemia. Cerebrovasc Dis 2000;110:165–9. [ Links ]
96. Ferrer I. Mecanismos de muerte neuronal en la isquemia cerebral. Rev Neurol 1999; 29 (6):515–21. [ Links ]
97. Wurtman RJ. La neurotoxicidad en la isquemia cerebral: del laboratorio a la clínica. Rev Neurol 1999; 29(6):524–26. [ Links ]
98. Murakami K, Kondo T, Chan PH. Reperfusion following focal cerebral ischemia alters distribution of neuronal cells with DNA fragmentation in mice. Brain Res 1997; 751:160–4. [ Links ]
99. Leist M, Volbracht C, Kühnle S, Fava E, Ferrando–May E. Caspase–mediated apoptosis in neuronal excitotoxicity triggered by nitric oxide. Mol Med 1997; 3(11):750–64. [ Links ]
100. Bonfoco E, Krainc D, Ankarcrona M, Nicoreta P, Lipton SA. Apostosis and necrosis: two distinct events induced, respectively by mild and intense insults with N–metyl–D–aspartate or nitric oxide/ superoxide in cortical cell cultures. Proc Nati Acad Sci USA 1995; 92(16):7162–6. [ Links ]
101. Du C, Hu R, Csernansky CA, Hsu CY, Choi D W. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab 1996; 16 (2): 195–201. [ Links ]
102. Endres M, Namura S; Shimizu–Sasamata M, Waeber C, Zhang L. Attenuation of delayed neuronal death after mild focal ischemia in mice by Inhibition of the caspase family. J Cereb Blood Flow Metab 1998; 18(3):238–47. [ Links ]
103. Fink K, Zhu I, Namura S, Shimizu–Sasamata M, Endres M. Prolonged therapeutic window for Ischemic brain damage caused by delayed caspase activation. J Cereb Blood Flow Metab 1998; 18(10):1071–6. [ Links ]

