










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkCiencias marinas
Print version ISSN 0185-3880
Cienc. mar vol.29 n.4 Ensenada Oct. 2003
Artículos
Mercury in sediments and pore waters at a contaminated site in the Tagus estuary
Mercurio en sedimentos y aguas intersticiales de un sitio contaminado del estuario del Tajo
João Canário1*, Carlos Vale1 and Miguel Caetano1
1 IPIMAR - Instituto de Investigação das Pescas e do Mar, Departamento de Ambiente Aquático - Lab. Metais Pesados, Av. Brasilia, 1449-006 Lisboa, Portugal. *E-mail: jcanario@ipimar.pt
Recibido en noviembre de 2000;
aceptado en octubre de 2002.
Abstract
Two sediment cores were collected at a contaminated site of the North Channel in the Tagus estuary. Sediments were sliced in 1 to 2-cm vertical sections. Major and minor elements, total organic carbon, total mercury (HNO3 digest), acid volatile sulphides -AVS- (1M HCl and 3M HCl) and simultaneously extractable iron, manganese and mercury were determined in the solids. Labile and total mercury (after UV irradiation) were measured in pore waters. Total mercury concentrations in the first 25 cm of sediment were around 100 nmol g-1, reflecting mainly anthropogenic discharges. A slightly compacted sediment layer (< 1 cm thickness) containing lower Hg concentrations (5 nmol g-1) capped the contaminated sediment. Mercury extracted with 1M HCl was only detected in this layer where AVS are low. In the sediment column, AVS were higher (50 to 197 µmol g-1), labile mercury was detected in pore waters down to 3.5-cm depth, and total dissolved Hg showed a broad maximum at 6 cm of depth (1.1 nM). The co-existence of high AVS and high total dissolved mercury implies the presence of ligands in pore waters with high affinity to mercury, which competes with sulphides and retains the metal in solution.
Key words: mercury, sediment, pore waters, Tagus estuary.
Resumen
Se colectaron dos núcleos de sedimento en un sitio contaminado del Canal del Norte del estuario del Tajo. Los sedimentos fueron cortados en secciones verticales de 1 a 2 cm. Se determinaron elementos mayores y menores, carbón orgánico, mercurio total (digeridos con HNO3), sulfuros volátiles en ácido -AVS- (HCl 1M y HCl 3M), así como hierro extraíble, manganeso y mercurio en los sólidos. El mercurio lábil y total (después de irradiarlo con UV) fue medido en aguas intersticiales. Las concentraciones totales de mercurio en los primeros 25 cm de profundidad del sedimento fueron de alrededor 100 nmol g-1, reflejando principalmente descargas antropogénicas. Una capa poco compactada de sedimento (< 1 cm de grosor) con bajas concentraciones de Hg (5 nmol g-1) cubría el sedimento contaminado. El mercurio extraído con HCl 1M sólo fue detectado en esta capa donde los AVS son bajos. En la columna de sedimento, los AVS fueron más elevados (50 a 197 µmol g-1). El mercurio lábil fue detectado en el agua intersticial hasta los 3.5 cm de profundidad y el Hg disuelto total mostró un amplio máximo a los 6 cm (1.1 nM). La coexistencia de una gran cantidad de AVS y un alto contenido de mercurio disuelto total implica la presencia de ligandos en las aguas intersticiales, con una alta afinidad al mercurio, los cuales compiten con los sulfuros y mantienen el metal en solución.
Palabras clave: mercurio, sedimento, agua intersticial, estuario del Tajo.
Introduction
The influence of diagenetic reactions on the remobilization of mercury in contaminated sediments is very important due to the well-known toxicity of mercury forms (Covelli et al., 1999). A large number of studies have been published on mercury distribution in sediments, water and organisms of contaminated areas (eg., Coquery et al., 1995; Pereira et al., 1998; Bjerregaard et al., 1999). These works showed that mercury is associated with the particulate phase matter, mainly due to its complexation with organic matter (Mantoura et al., 1978) and association with sulphur compounds (Drobner et al., 1990; Morse and Luther, 1999). In several cases, it was possible to relate the vertical profiles of total mercury with the historical evolution of mercury contamination in the area (Gobeil and Cossa, 1993; Pereira et al., 1998). Only a few studies have addressed the diagenetic behaviour of mercury and its mobility in coastal sediments (Bothner et al., 1980; Gobeil and Cossa, 1993; Gagnon et al., 1997). By determining the distribution and speciation of mercury in both the dissolved and solid phases, these investigations have shown that mercury seems to be bond to organic matter, a minor fraction being recycled with the Mn and/or Fe-oxides near the redox boundary, and some being adsorbed to or coprecipitated with acid volatile sulphides (AVS).
The North Channel of the Tagus estuary has been a site of industrial discharges for several decades (Ferreira, 1995), resulting in particularly high levels of mercury in surface sediments, suspended matter and water (Figuères et al., 1985). This paper reports the vertical distribution of mercury in sediments and pore waters nearby the anthropogenic source and examines the retention and possible mobility within sediments.
Material and methods
Study site
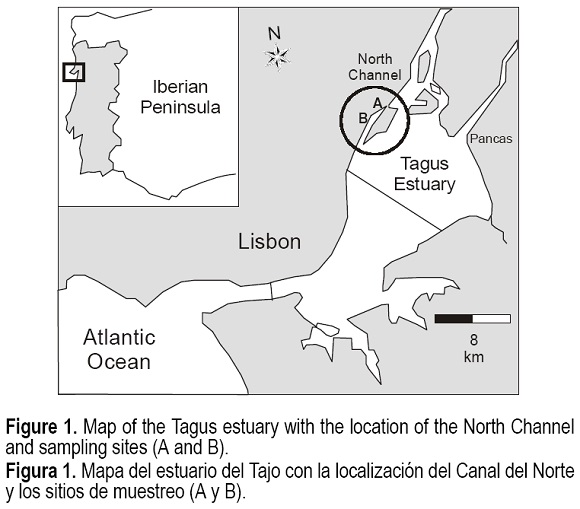
The study site is located in the North Channel of the Tagus estuary (fig. 1). The channel is 14 km long, approximately 200 m wide, and a maximum water column depth of 2 m. Salinity varies in a broad interval with the tide. Under moderate river flow, salinity may change from 2 PSU at low tide in upstream zones to 32 PSU at high tide in downstream areas (Canário, unpublished data). The low hydrodynamics of the channel favors the deposition of large quantities of suspended particulate matter (Vale, 1990), the mean deposition rate being estimated in 0.8 cm yr-1 (Nunes, 1993).
Sampling
Two sediment cores were collected in the intertidal zone nearby the major anthropogenic emission of Hg at the North Channel in July 1999 (core A) and January 2000 (core B). Each core was sampled by hand with a 10-cm diameter PVC tube. Temperature, redox potential (EH) and pH were measured at regular intervals along the cores. The cores were rapidly sliced in situ in 0-0.5-cm thickness (topmost layer), 1-cm intervals down to a 4-cm depth and, below this, they were sectioned in 2-cm slices. Samples were stored in leak-proof tubes that were completely filled up and sealed in order to avoid oxidation by air. These operations did not exceeded 10 minutes per sample, which has showed not to cause significant changes on sulphur speciation in solids (Madureira et al., 1997). Each sample was stored in two different vials, one for determination of water content, total organic carbon (Corg), total metal concentrations, AVS and simultaneously extracted Fe, Mn and Hg, and a second portion for pore water analysis. For this purpose, leak-proof tubes were filled up with sediment to avoid air bubbles and centrifuged at 6000 rpm for 10 min (4°C). The supernatant was filtered through 0.45-µm polycarbonate membranes and acidified to pH < 2 with concentrate HNO3. A small quantity of each sediment layer was frozen for AVS analysis, and the remaining was oven-dried at 40°C, disaggregated, homogenised and stored in polyethylene bottles until the analysis.
Measurements in situ
Redox potential was measured using a platinum combined redox electrode (Ingold) calibrated with a 220 ± 5mV solution (vs. Ag/AgCl, 25°C). Measurements were converted to normal hydrogen potential. The pH determinations were done with a glass-combined electrode (Mettler) calibrated with pH 4.00 and pH 7.00 ± 0.02 buffers (20°C). The oxygen penetration depth was measured previously before slicing by polarography using a needle electrode (Diamond Electro-Tech Inc.) as described by Brotas et al. (1990).
Water content and total organic carbon
Water content was determined by weight loss at 105°C. Total organic carbon content in dry sediment samples was determined using a Carlo-Erba Mod. 1106 analyser after preparing the sample according to Byers et al. (1978).
Elemental analysis of sediments
Total determinations of Al, Si, Ca, Mg, Fe and Mn were done by mineralising the sediment samples with a mixture of acids (HF, HNO3, HCl and H3BO3) according to the method described by Rantala and Loring (1977). The solutions obtained were then analysed by flame atomic absorption spectroscopy (Perkin Elmer AAnalyst 100) using direct aspiration into an N2O-acetylene flame (Al, Si, Ca and Mg) or air-acetylene flame (Fe and Mn). For total mercury determinations -100 mg of sediment were digested overnight at room temperature with 50 cm3 of 4M HNO3 in borosilicate glass erlenmeyers. Solutions were heated in a sand bath at 60-70°C for two hours, transferred to 100 cm3 volumetric flasks and the volume completed with 1.5% HNO3 (Pereira et al., 1998). Mercury was determined by cold vapour atomic absorption spectrometry in a Perkin-Elmer Flow Injection Mercury System FIAS-100 using SnCl22H2O (1.1%) solution in HCl (3%), as reducing agent. Detection limits for total metal analysis were 0.3 nmol g-1 for Hg, 41 µmol g-1 for Al, 11 µmol g-1 for Si, 25 nmol g-1 for Fe and 0.21 µmol g-1 for Mn. Precision errors were: Hg 1.0%, Al 2.3%, Si 1.6%, Ca 4.7%, Mg 3.6%, Fe 2.3% and Mn 1.0% (P < 0.05). International certified standards (MESS-2 and PACS-1 for Hg, and AGV-1, GSP-1, G-2, MESS-1 and BCSS-2 for the other elements) were used to control the accuracy of our procedure. For all metals investigated, obtained and certified values were not statistically different (P < 0.01).
Determinations of AVS and associated metals
Approximately 50 mg of wet sediment samples were used for AVS determinations. Sulphides from AVS were released in 1 and 3M HCl, purging the system with nitrogen gas, and trapping the H2S in 1 M NaOH solution (Henneke et al., 1991; Madureira et al., 1997). Hydrogen sulphide trapped in the basic solution was determined by voltametric methods (Luther et al., 1985) using a Methrom device equipped with a 693 VA processor and a 694 VA stand. The recovery of standard sulphide solution was 97% and detection limit of the method was 0.01 nM. The simultaneously extracted Fe, Mn and Hg released from AVS by acidification of the sediment with two acid solutions (1M and 3M HCl) were analysed as described above. Detection limits for Fe, Mn and Hg extracted with the AVS were 0.14 µmol g-1, 0.36 µmol g-1 and 0.01 nmol g-1, and precision errors were 2.0, 3.0 and 3.2% (P < 0.05), respectively.
Determinations of labile and total mercury in pore waters
Labile mercury (easily reduced by a SnCl2 solution) was measured directly from the filtered solutions by cold vapour atomic fluorescence spectroscopy using a cold vapour generator (PSA, model 10.003) embeded to a fluorescence detector (PSA Merlin 10.023). Total dissolved mercury was measured in the same equipment after UV oxidation with a 1000-W UV lamp following the method described by Mucci et al. (1995) and Montgomery et al. (1995). Accuracy and precision were determined by preparing different stock solutions of 0.010, 0.025, 0.050, 0.010, 0.015 and 0.025 nM (Hg2+) from 1000 ppm Merck stock solution in 2% HNO3. The different solutions were always prepared and measured between 2 samples. The detection limit and the precision error were 0.01 nM and 4.0%, respectively (P < 0.05).
Glassware, sample containers and reagents
All labware used in this work was of borosilicate glass, polycarbonate or Nalgene brand F.E.P. which had been cleaned by the following procedure: 24h in 20% Extran™ (Milli-Q water) and 48h in 25% HNO3 (Milli-Q water) followed by several washes with Milli-Q water. The polycarbonate membranes were cleaned in 25% HNO3 for 3 days and then several washes with Milli-Q water. All the material was dried in a clean room and kept in plastic bags or Petri dishes (polycarbonate membranes) until analysis. Reagents used in this work were Merck suprapur™ grade and, in the case of mercury analysis, mercury-free grade.
Results
Water content, pH, EH and O2
Visual inspection of the collected cores indicated that all sediment layers were constituted by fine particles and no buried animals were detected. The first 2-3 cm of cores A and B contained approximately 60% of water, decreasing abruptly with depth to 42% and then remained constant. Values of pH decreased gradually from 8.6 at the sediment-water interface to 7.9 at 20-cm depth and then remained relatively constant. The redox potential (EH) was -70 mV at the surface, around -170 mV in the first 10 cm, and between -210 and -250 mV at deeper layers. Oxygen levels in overlying water were around 240 mM and its concentrations decreased sharply in the first millimetre of the sediment (121 mM) becoming undetectable below the 2-mm depth.
Major elemental composition of sediments
Total organic carbon ranged from 0.3 to 2.1 mmol g-1 with higher levels in the upper sediment layers (fig. 2). Concentrations of major and minor elements varied in the following ranges: Al from 3.1 to 5.1 mmol g-1, Si from 2.7-4.5 mmol g-1, Ca 2.1-3.7 mmolg-1, Mg 0.45-1.25 mmolg-1, Fe 0.50-0.71 mmol g-1 and Mn 8.0-12 µmol g-1. Aluminium concentrations and Si/Al ratios were characteristic of fine sediments. Concentrations of Ca and Mg in the upper layers were high in comparison with sediments from other areas of the Tagus estuary (Vale, 1990; Vale et al., 1999). The vertical distribution of Fe/Al ratios (fig. 2) showed higher values in the upper 7-cm layer (max. 0.23 on molar basis in core A). Below this depth, Fe/Al ratios decreased to 0.11 and remained relatively constant until the end of the core. A lower variability of the Fe/Al ratio was found in core B. The vertical profile of Mn/Al ratios was similar in both cores, showing higher values (3.2) at the topmost layers, and decreasing to 2.0 in the first 5-cm depth.
Total mercury in sediments
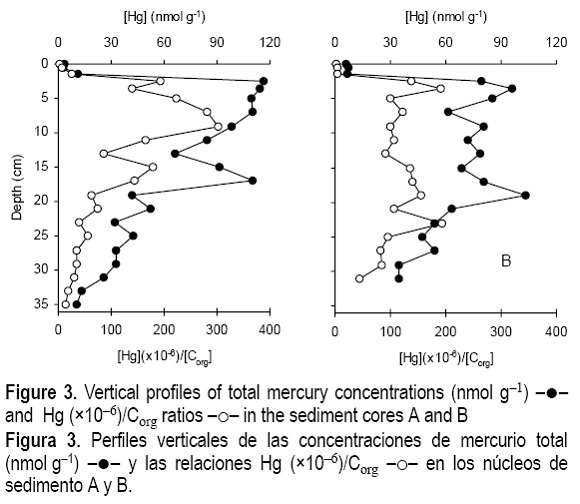
The total mercury concentration in the sediment solids exhibited similar vertical profiles in both cores A and B (fig. 3). Upper layers (with higher water content) had approximately 3 nmol g-1 and at 2.5-cm depth mercury concentrations increased abruptly to more than 115 nmolg-1. Then, levels decreased with depth but another enhancement was found around 18 cm in both cores. Below this depth levels decreased gradually, although at 36-cm depth concentrations were still 34.7 nmol g-1 (core B) and 10.3 nmol g-1 (core A), which are far above the baseline values (0.25 nmol g-1) generally observed in coastal sediments (Pereira et al., 1998). When mercury concentrations are normalized to Corg, the sharp variation in the upper sediment layers remains the same (fig. 3), but the second concentration peak of core A was attenuated and, in core B, it was suppressed.
AVS and simultaneously extracted metals
The vertical profile of AVS in core A exhibited low values in the topmost layers, a pronounced increase to 150 nmol g-1 at 2.5 cm, and then successive maxima and minima (fig. 3). Manganese simultaneously extracted with 1M HCl ranged in a narrow interval (0.3-0.5 nmol g-1) except in the topmost layer where it reached 2.3 nmol g-1. Iron ranged between 33 and 43 nmol g-1. Mercury concentrations were uniformly low (< 0.01 nmol g-1), except for the topmost layer (0-0.5 cm) where extracted mercury reached 0.02 nmol g-1, corresponding to 0.7% of total mercury. The fraction of mercury extracted with the 3 M HCl solution in three selected sediment layers (1-2 cm, 16-18 cm, and 26-28 cm) increased considerably, reaching 2.95 nmol g-1, 18.8 nmol g-1 and 5.45 nmol g-1, respectively. These values correspond to 17-27% of the total mercury. Iron and Mn concentrations increased approximately 10 times relatively to 1M HCl extractions. The substantial increases of the three metals analysed are unlikely to be attributable to dissolution of sulphur compounds since AVS values in the two acid extractions did not differ by more than 15%.
Mercury in pore waters
Vertical profiles of labile mercury and total mercury in pore waters of core A are shown in figure 4. Labile mercury concentrations were less than 5% of the total dissolved mercury and were only detected in the first 3-cm depth, reaching 0.031 nM. Deeper in the sediments, levels were lower than 0.01 nM. The vertical profile of total dissolved mercury showed a broader peak, with concentrations higher than 1.1 nM between 1 and 10 cm. Below this depth, levels were lower than 0.125 nM.
Discussion
The total mercury concentration in the North Channel sediments of the Tagus estuary, 100 nmol g-1, reflect the industrial discharges that have occurred during the last few decades in the area. These results are in line with previous works that report Hg contamination of surface sediments, suspended matter and water (Figuères et al., 1985; Vale et al., 1999; Simas, 1999). Mercury concentrations in the upper 2-cm layers were lower, approximately 5 nmol g-1. These values are comparable to levels observed in suspended particulate matter (Simas, 1999), and lower than concentrations reported for samples collected in 1981 (Figuères et al., 1985), before the reduction of the industrial mercury discharges (Simas, 1998). Consequently, the severely contaminated sediments are, at present, covered with a layer of less contaminated particles. However, this natural capping effect may be repeatedly interrupted by the resuspension of bottom sediments in periods of stronger tidal currents (Vale and Sundby, 1987), as suggests the low compaction of the topmost sediment layers.
The question of great environmental importance is whether diagenetic processes lead to a substantial post-depositional redistribution of the anthropogenic mercury. The vertical profiles of labile and total dissolved mercury indicate that partitioning of mercury between pore waters and sediment solids varied with depth. It is suggested that these changes resulted from the diagenetic remobilization that occurred mainly in the upper sediment layers. Nevertheless, by comparing the total concentrations in solution (order of nM) and solids (hundreds of nmol g-1), one may conclude that dissolved Hg accounts for less than 1% of the bulk sediment Hg content. The low fraction of Hg in pore waters is similar to that reported in other contaminated coastal environments (eg., Gobeil and Cossa, 1993). In addition, there is a lack of covariance between Hg in the dissolved and solid phases. Despite only one core has been analysed for pore waters, the results suggest that pore water Hg concentrations are not simply controlled by an equilibrium exchange between the two phases. The fact that labile mercury decreased between the 2nd and 3rd centimetres while AVS increased may indicate that the most labile forms are removed by the precipitation of amorphous sulphides. It is not surprising that the amount removed was not detected in the simultaneously extracted fraction of mercury (< 0.03 nM) due to the low quantity of labile mercury. The co-existence of a broad maximum of total dissolved mercury with high AVS (fig. 3) indicates that these soluble mercury forms were not removed substantially by the precipitation of sulphides. Although further investigation should be done with respect to the ligands responsible for keeping mercury in solution, it may be concluded that those ligands have a strong affinity to mercury in order to compete with the dissolved hydrogen sulphides. Possible candidates are humic substances, due to the large number of functional groups (Mantoura et al., 1978) and polysulphides that form soluble compounds with mercury (Lindberg and Harriss, 1974), and consequently depress the activity coefficient of Hg2+ (αHg2+). The low αHg2+ might explain the small amount of mercury associated with AVS, accounting for less than 0.3% of the total mercury in the solids. Studies in other contaminated environments report higher contributions of Hg-FeS compounds to the total mercury concentrations (Gagnon et al., 1997). Using a stronger acid solution (3M HCl) about 20% of mercury was removed from the solids. Since sulphides (extracted with 3M HCl) did not varied substantially in relation to the weaker extraction (1M HCl), mercury incorporated as other forms rather than sulphides is removed. In spite of the well documented association between mercury and particulate organic matter (Baldi and Bargagli, 1984; Mason and Lawrence, 1999) no correlation was found in the samples analysed from cores A and B.
In both cores there is a broad increment of Hg around 18 cm (fig. 3). This may be interpreted as an increment in the industrial discharge, a high organic matter content in these layers (Mason and Lawrence, 1999), or diagenetic precipitation. In core B, the normalization of Hg to Corg suppresses this peak, indicating that Hg increment is due to the higher organic matter content of these layers. In core A the increment remained after normalization to Corg. Furthermore, the decrease of total dissolved mercury in pore waters below 6 cm coincides with the concentration enhancement of total mercury in solids. If these alterations were attributed to the diagenetic mobilisation due to the steeper concentration gradient of total mercury in pore water, the diffusive flux, J, could be estimated by (Berner, 1980):
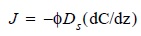
where Φ is the porosity, Ds is the diffusion coefficient, and dC/dz is the concentration gradient. If we assume Ds to be ΦDo (Ullman and Aller, 1982), where Do is the molecular diffusion coefficient for mercury in seawater, and taking Do = 6.09 x 10-6 cm2 s-1 (Boudreau, 1997), then the downward flux would be J = 1.38 pmol dm 2 d-1. Based on the vertical profile of total mercury, the concentration peak at ~18 cm corresponds to 21 nmol dm-2. Assuming 0.8 cm yr1 for the sedimentation rate in this area (Nunes, 1993) we can conclude that the annual contribution of the diffusive flux is approximately 2%. This negligible quantity suggests that the enhancement of total mercury in the solids is related to an episodic discharge from local industries, which is in agreement with the high Hg/Corg ratio. Due to its low mobility in the sediments, mercury profiles seem to document the historical evolution of mercury contamination in the North Channel of the Tagus estuary, as proposed by several authors for many aquatic systems (Klein and Goldberg, 1970; Aston et al., 1973; Breteler et al., 1984; Pereira et al., 1998).
Conclusions
The results of this study indicate that mercury profiles in the sediments probably reflect the temporal changes in the rate at which mercury has been poured to the North Channel of the Tagus estuary. The high total mercury concentrations in the first 25 cm of sediments reflect anthropogenic discharges. Only the low-compacted sediment layer blanking the contaminated sediments contained lower Hg concentrations. In the sediment column AVS was high, labile mercury in pore water was low, and the total dissolved Hg showed a broad maximum at 6 cm. The co-existence of high AVS and mercury in pore waters indicates the presence of ligands with a great affinity to mercury. The formation of soluble mercury compounds in pore waters seems to limit the adsorption and coprecipitation onto AVS.
Acknowlegements
The authors thank the colleagues Eduarda Pereira, Elsa Ramalhosa and Maria-João Madureira for their help on the analytical work.
References
Aston, S.R., Bruty, D., Chester, R., and Padgham, R.C. (1973). Mercury in lake sediments: a possible indicator of technological growth. Nature, 241: 450-451. [ Links ]
Baldi, F., and Bargagli, R.. (1984). Mercury pollution in marine sediment near a chlor-alkali plant: Distribution and availability of the metal. Sci. Total Environ., 39: 15-26 [ Links ]
Bjerregaard, P., Andersen, B., and Rankin, J.C. (1999). Retention of methyl-mercury and inorganic mercury in rainbow trout Oncorhynchus mykiss (W): effect of dietay selenium. Aquat. Toxicol., 45: 171-180. [ Links ]
Berner, R. (1980). Early Diagenesis. A Theorical Approach. Princeton University Press, USA, 241 pp. [ Links ]
Bothner, S.S., Jahnke, R.A., Peterson, M.L., and Carpenter, R. (1980). Rate of mercury loss from contaminated estuarine sediments. Geochim. Cosmochim. Acta., 44: 273-285. [ Links ]
Boudreau, B.P. (1997). Diagenetic Models and their Impletation: Modelling Transport and Reactions in Aquatic Sediments. Springer-Verlag, New York, 441 pp. [ Links ]
Breteler, R.J., Bowen, V.T., Schneider, D.L., and Henderson, R. (1984). Sedimentological reconstruction of recent pattern of mercury pollution in the Niagara River. Environ. Sci. Tech., 18: 404-409. [ Links ]
Brotas, V., Amorim-Ferreira, A., Vale, C., and Catarino, F. (1990). Oxygen profiles in intertidal sediments of Ria Formosa (S. Portugal). Hydrobiologia. 207: 123-129. [ Links ]
Byers, S C., Mills, E.L., and Stewart, L. (1978). A comparision metods of determinig organic carbon in marine sediments, with suggestions for a standard method. Hydrobiologia, 58: 43-47. [ Links ]
Coquery, M., Cossa, D., and Martin, J.M. (1995). The distribution of dissolved and particulate mercury in three Siberian Estuaries and adjacent artic coastal waters. Water, Air, Soil Pollut., 80: 653-664. [ Links ]
Covelli, S., Faganeli, J., Horvat, M., and Brambati, A. (1999). Porewater distribution and benthic flux measurements of mercury and methylmercury in the Gulf of Trieste. Estuar. Coast. Shelf Sci., 48: 415-428. [ Links ]
Drobner, E., Huber, H., Wachterhauser, G., Rose, D., and Steller, K. (1990). Pyrite formation linked with hydrogen sulphide evolution under anaerobic conditions. Nature, 546: 742-744. [ Links ]
Ferreira, J.G. (1995). An object-oriented ecological model for aquatic ecosystem's. Ecol. Model., 79: 21-34. [ Links ]
Figuères, G., Martin, J.M., Meybeck, M., and Seyler, P. (1985). A comparative study of mercury contamination in the Tagus Estuary (Portugal) and major French Estuaries (Gironde, Loire, Rhône). Estuar. Coast. Shelf Sci., 20: 183-203. [ Links ]
Gagnon, C., Pelletier, É., and Mucci, A. (1997). Behaviour of anthropogenic mercury in coastal marine sediments. Mar. Chem., 59: 159-176. [ Links ]
Gobeil, C., and Cossa, D. (1993). Mercury in sediments and sediment pore water in the Laurentian Trough. Can. J. Fish. Aquat. Sci., 50: 1794-1800. [ Links ]
Henneke, E., Luther, G.W., and De Lange, G.J. (1991). Determination of inorganic sulphur speciation with polarographic techniques: some preliminary results from recent hypersaline anoxic sediments. Mar. Geol., 100: 115-123. [ Links ]
Klein, D.H., and Goldberg, E.D. (1970). Mercury in the marine environment. Environ. Sci. Tech., 4: 765-768. [ Links ]
Lindberg, S., and Harriss, R.C. (1974). Mercury-organic matter associations in estuarine sediments and interstitial water. Environ. Sci. Technol., 8: 45 9-462. [ Links ]
Luther, G. W., Giblin, A. E. and Varsolona, R. (1985). Polarographic analysis of sulphur species in marine porewaters. Limnol. Oceanogr., 30: 727-736. [ Links ]
Mantoura, R.F.C., Dickson, A., and Riley, J.P. (1978). The complexation of metals with humic materials in natural waters. Estuar. Coast. Mar. Sci., 6: 387-408. [ Links ]
Madureira, M.J., Vale, C., and Simões Gonçalves, M.L. (1997). Effect of plants on sulphur geochemistry in the Tagus salt-marshes sediments. Mar. Chem., 58: 27-37. [ Links ]
Mason, R.P., and Lawrence, A.L., (1999). Concentration, distribution, and bioavailability of mercury and methylmercury in sediments of Baltimore Harbor and Chesapeake Bay, Maryland, USA. Environ. Toxicol. Chem., 18 (11): 2438-2449. [ Links ]
Montgomery, S., Mucci, A., Lucotte, M., and Pichet, P. (1995). Total dissolved mercury in the water column of several natural and artificial aquatic systems of Northern Quebec. Can. J. Fish. Aquat. Sci., 52: 2483-2492. [ Links ]
Morse, J.W., and Luther, G.W. (1999). Chemical influences on trace metal-sulphide interactions in anoxic sediments. Geochim. Cosmochi. Acta., 63: 3373-3378. [ Links ]
Mucci, A., Lucotte, M., Montgomery, S., Plourde, Y., Pichet, P., and Tra, N. (1995). Mercury remobilization from flooded soils in a hydroelectric reservoir of Northern Quebec. La Grande-2. Results of a soil resuspension experiment. Can. J. Fish. Aquat. Sci., 52: 2507-2517. [ Links ]
Nunes, L.M. (1993). Distribuição de Metais Pesados nos Sedimentos da Cala do Norte do Estuário do Tejo. ECOTEJO, Rel. A-8402-01-93-UNL, Ed. DCEA/FCT, 25 pp. [ Links ]
Pereira, M.E., Duarte, A.C., Millward, G.E., Abreu, S.N., and Vale, C. (1998). An estimation of industrial mercury stored in sediments of a confined area of the lagoon of Aveiro (Portugal). Water Sci. Technol., 36(6-7): 125-130. [ Links ]
Rantala, R. T. T., and Loring, D. H. (1977). A rapid determination of 10 elements in marine suspended particulate matter by atomic absorption. Atom. Absorpt. Newsllet., 16: 51-52. [ Links ]
Simas, M. T. (1998). Contaminação por Mercúrio em Alguns Níveis Tróficos da Cala do Norte do Estuário do Tejo. MSc. Thesis, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 121 pp. [ Links ]
Simas, M.T. (1999). Mercúrio na matéria particulada e nos sedimentos da Cala do Norte do Estuário do Tejo. ECOTEJO, Rel. A-8503-02-99-UNL, Ed. DCEA/FCT, 25 pp. [ Links ]
Ullman, W.J., and Aller, R.C. (1982). Diffusion coefficients in nearshore marine sediments. Limnol. Oceanogr., 27: 552-556. [ Links ]
Vale, C., and Sundby, B. (1987). Suspended sediment fluctuations in the tagus on semi-diurnal and fortnightly time scales. Estuar. Coast. Shelf Sci., 25: 495-508. [ Links ]
Vale, C. (1990). Temporal variations of particulate metals in the Tagus River Estuary. Sci. Total Environ., 97/98: 137-154 [ Links ]
Vale, C., Quintans, M., Raimundo, J., Santos, I., Brito, P., Fonseca, N., and Santos, J. (1999). Ponte Vasco da Gama - Relatório de Progresso de Monitorização Ambiental da Fase de Exploração -Janeiro 1999, 13 pp. [ Links ]

