










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkCiencias marinas
Print version ISSN 0185-3880
Cienc. mar vol.29 n.4 Ensenada Oct. 2003
Artículos
Iridio: Una opción para medir el potencial redox
Iridium: An option for measurement of the redox potential
Lorena M. Ríos-Mendoza1,2*, J. Vinicio Macías-Zamora2 y Alberto R. Zirino-Weiss3
1 Facultad de Ciencias Marinas.
2 Instituto de Investigaciones Oceanológicas, Universidad Autónoma de Baja California, Apartado postal 453, Ensenada, CP 22800, Baja California, México. *E-mail: lrios@uabc.mx
3 Naval Command, Control and Ocean Surveillance Center, RDT&E Div.,Code D361, San Diego, CA 92152, USA.
Recibido en octubre de 2001;
aceptado en diciembre de 2002.
Resumen
Se presentan los resultados relevantes en la búsqueda de un electrodo alternativo al platino para mediciones redox. Para estos ensayos se usó un grupo de cinco electrodos inorgánicos inertes (platino, grafito, carbón vitrificado, oro e iridio). Nuestros resultados, respaldados con cálculos de modelos termodinámicos, utilizando el programa MINEQL+ sugieren que, a diferencia de otros estudios, la pareja O2/H2O2 es la que controla el potencial redox bajo las condiciones experimentales llevadas a cabo en este trabajo. En aguas bien aireadas (PO2 = 0.21 atm) y con pH = 8, el valor del EH = 0.748 V y del pE en la superficie es de 12.67 (pE = (F/2.3RT) EH = 12.67). Sin embargo, en la práctica, el potencial medido experimentalmente, tanto por el electrodo de Pt como por el de Ir, es de entre +0.45 a +0.50 V. Adicionalmente, la presencia de H2O2 fue deducida a partir de nuestras mediciones y su concentración fué estimada en valores cercanos a 10-8 M, coincidiendo con valores reportados por otros autores para aguas naturales. Las mediciones a que se sometieron los electrodos de Pt y de Ir fueron: variaciones de O2 manteniendo el pH constante y viceversa; mediciones de algunos pares redox comunes en agua natural pero medidos en agua de mar sintética (se incluyeron Fe2+/Fe3+, NO3-/NO2- y, I3-/I-); mediciones en medio natural en planicies lodosas de San Quintín, Baja California; y finalmente se hicieron mediciones de potencial redox en agua de mar natural sin filtrar y en agua de mar natural filtrada y oxidada mediante luz UV. Los resultados muestran que las respuesta más cercanas a las predichas por un modelo termodinámico se obtienen con el electrodo de Ir. El electrodo de Pt en general pareció responder de manera mas lenta que el Ir.
Palabras clave: potencial redox, iridio, electrodo redox, platino.
Abstract
Some relevant results are presented of research leading to an alternative redox electrode to platinum. Several different electrodes were subjected to experiments under controlled conditions, mainly in artificial seawater. Among those tested were platinum, graphite, glassy carbon, gold and iridium. Our results, when combined with those estimated by using a model based on the MINEQL+ program, suggest that the O2/H2O2 redox couple is the one that controls the redox potential under these experimental conditions. In well-oxygenated waters (PO2 = 0.21 atm) and pH = 8, the value calculated for EH = 0.748 V and the pE at the surface should read 12.67 (pE = (F/2.3RT) EH = 12.67). However, in practice, the redox potential experimentally measured was only between +0.45 and +0.50 V. Additionally, the presence of H2O2 was determined from our own measurements. Its concentration was estimated at about 10-8 M. This value is in good agreement with values measured and reported by other authors for surface waters under natural conditions. The four sets of measurements that were carried out were: measurements where the O2 concentration of the solution was changed and the pH was kept constant (and vice versa); measurements of several redox couples (including Fe2+/Fe3+, NO3-/NO2- and I3-/I-); measurements in mud flats at San Quintín, Baja California; and measurement of the redox potential in natural seawater and in filtered and UV seawater. Most measurements indicate that the electrode with a response closest to that predicted by thermodynamics is the Ir electrode. We also found that the reaction of Pt, when compared to Ir, appeared to be slow.
Key words: redox potential, iridium, redox electrode, platinum.
Introducción
Existen dos componentes fundamentales del medio ambiente natural, protones y electrones. La concentración o actividad de los protones en el sistema acuoso natural indica el grado de acidez o basicidad que posee un medio. Usualmente ésta se expresa en una escala de pH (pH = -log aH+), en cambio, el potencial redox (pE= -log ae-) es solo una expresión de la tendencia de la reversibilidad del sistema redox a ser oxidado o reducido (Zobell, 1946; Sillén, 1952; Baas-Becking et al., 1960; Bates, 1964; Thorstenson, 1984).
Los estudios del pE y el pH son básicos para la comprensión de los sistemas acuosos, ya que juntos reflejan la especiación de diferentes elementos que, a su vez, directa o indirectamente afectan los procesos químicos en el medio ambiente natural (Langmuir, 1997). En los sistemas oceánicos, los procesos biogeoquímicos se encuentran fuertemente influenciados por las reacciones redox, sin embargo, la mayoría de estas reacciones son de cinética muy lenta sin mediadores catalíticos y, en algunas ocasiones, no se llegan a obtener verdaderos equilibrios redox. Afortunadamente existen mediadores biológicos que tienden a crear un seudo equilibrio que puede considerarse como próximo a un estado estacionario y, a partir de este, hacer aproximaciones de equilibrio que puedan ser utilizadas en los cálculos de especiación redox (Chester, 1990; Barcelona y Holm, 1991; Parsons, 1978).
En los sedimentos el potencial redox es el factor más importante que determina la estabilidad y la transformación bioquímica de la materia orgánica, así como la distribución, tipo y actividad fisiológica de las bacterias y otros microorganismos que se encuentran en los sedimentos (Teasdale et al., 1998)
De la misma manera, dentro de las células la energía redox es transportada mediante la transferencia de electrones a través de moléculas llamadas acarreadoras de electrones, siendo los organismos fotosintéticos los que usan la energía solar para sintetizar moléculas que son termodinámicamente inestables y los organismos no-fotosintéticos tienden a transformar estos compuestos a formas estables mediante reacciones redox.
Existe un gran número de publicaciones sobre mediciones redox en sistemas acuosos naturales. Sin embargo, los valores obtenidos mediante estas mediciones no han sido fáciles de interpretar, y no se ha podido explicar si el pE medido caracteriza a todo el sistema redox (Mansfield, 1925; Morris y Stumm, 1967; Whitfield, 1969, 1974; Stumm, 1978; Champ et al., 1979; Bricker, 1982; Peiffer et al., 1992). Hacer entonces interpretaciones termodinámicas cuantitativas del pE equivaldria a conocer todos los pares redox efectivos al sensor y sus respectivas concentraciones, lo que resultaría totalmente impráctico.
Afortunadamente en ambientes acuosos naturales el número de pares redox que contribuyen al potencial redox es limitado y se le relaciona principalmente con Fe3+/Fe2+, Mn4+/ Mn2+, SO4=/HS-, O2/H2O2, O2/H2O, CO2/CH4, así como algunas sustancias orgánicas complejas que pueden contribuir al potencial (Morris y Stumm, 1967; Pettine, 2000). Es claro que las reacciones redox en aguas naturales no están en equilibrio, así que las mediciones del potencial redox que se realicen en el campo y las teóricas basadas en la termodinámica van a ser diferentes entre sí. Dependiendo de las condiciones físico-químicas de los diferentes pares redox que empiezan a dominar el sistema se determina un potencial eléctrico que puede ser medido mediante un electrodo.
En la actualidad el electrodo de platino es el más utilizado para las mediciones de potencial redox. Este electrodo presenta adsorción de sustancias tanto electroactivas como no electroactivas. Este efecto influye en las características del intercambio de las especies redox disueltas evitando que actúe como un electrodo inerte (Morris y Stumm, 1967; Whitfield, 1969, 1974; Champ, 1979; Vershinin y Rozanov, 1983; Peiffer et al., 1992). Este sensor, en sistemas bién aireados, actúa de manera análoga a la que se predice para un electrodo de Pt0/Pt-O (Whitfield, 1974), cuyo potencial es EH = 0.88 - 0.059 pH.
Esto sugiere que el electrodo de platino se comporta como un electrodo de pH en aguas bién oxigenadas, por lo que no responde a los cambios de potencial redox en el sistema.
El objetivo de este trabajo es presentar los resultados relevantes en la búsqueda de un electrodo alternativo al platino para mediciones redox. Presentamos al iridio como un sensor redox reproducible para mediciones en soluciones salinas óxicas. Se mostrará que éste muestra una respuesta superior al electrodo de platino y a otros electrodos redox frecuentemente recomendados en la literatura. Entre sus características notables resalta el ser un electrodo inorgánico inerte y de rápida respuesta electroquímica a los cambios de potenciales redox que ocurren en el sistema.
Consideraciones teóricas para el cálculo del potencial redox
Las reacciones redox involucran un cambio neto en el estado de oxidación formal de los elementos involucrados en una reacción. Las especies reductoras son capaces de donar electrones y las especies oxidantes son capaces de aceptar electrones. Los electrones libres no pueden existir en soluciones acuosas por lo que toda oxidación debe ser acompañada de su correspondiente reducción y viceversa (Domenico y Schwartz, 1998).
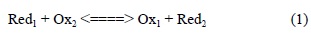
donde Red y Ox, se refieren a las formas reducidas y oxidadas, respectivamente. La transferencia de electrones, induce cambios en la energía eléctrica, produciéndose una diferencia de potencial (Eeq) que se mide con un electrodo redox. La relación termidinámica entre el potencial redox y la composición de la solución se expresa mediante la ecuación de Nernst, usando un potencial estándar (E0eq).

donde Eeqj es el potencial de electrodo en volts, que es medido contra el electrodo estándar de hidrógeno (SHE) comúnmente denotado como EH; E0eqj es el potencial redox estándar de la reacción (volts); R es la constante de gases (1.987 cal/mol °K); T es la temperatura absoluta (°K); F es la constante de Faraday (9.649 x104 C/mol); y nj es el número de electrones. El valor de 2.303 RT/F, a 25°C, es de 0.059V
Considerando al electrón como un reactivo participante de la reacción (1), se define la actividad del mismo en equilibrio como (Sillén, 1967; Truesdell, 1968; Morel y Hering, 1993)

donde ae- es la actividad del electrón y Kj es la constante de equilibrio. Esta actividad del electrón usualmente se expresa en cualquiera de las dos escalas, ya sea de pE ó EH. Estas dos escalas se relacionan entre sí de la siguiente manera:
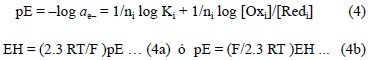
lo que significa que el pE puede describirse en términos de un potencial.
Baas-Becking et al. (1960) propusieron limitar la química de las aguas naturales en términos de pH y potencial redox por dos fronteras. El agua misma es oxidada por el oxígeno a altos pE y es reducida por el hidrogéno pE bajos. Sin embargo, las mediciones del EH en aguas naturales oxigenadas difieren de la frontera del O2/H2O. Esto, en parte puede reflejar la lentitud de la cinética en la oxidación del agua y la dificultad de medirla con un sensor redox (Breck, 1974; Williams, 1990).
Sato (1960) propone, de manera alternativa, que la pareja O2/H2O2 es la que controla el potencial redox. Esta diferencia es importante y se explica como sigue: el potencial de oxidación al cual el O2 es reducido a H2O esta dado por la siguiente ecuación:

En aguas bien aireadas (PO2 = 0.21 atm) y con pH = 8, el valor del EH es 0.748 V y el pE en la superficie, de acuerdo con la ecuación (4b), es de 12.67:
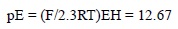
Sin embargo, en la práctica, mediciones experimentales muestran que no se alcanza dicho potencial. El potencial medido experimentalmente es tan sólo de entre +0.45 a +0.50 V (Cooper, 1937; Breck, 1972; Pettine y Millero, 1990; Silver, 1991; Teasdale et al., 1998). La explicación que se ofrece es la lentitud de la cinética para el rompimiento del enlace del O2 en la formación del H2O. La adquisición del segundo electrón del H2O2 por el H2O es muy lenta (Breck, 1972; Morel y Hering, 1993). Sato y Mooney (1960) hicieron mediciones del potencial redox en agua y propusieron el siguiente mecanismo de control del potencial redox en aguas oxigenadas;

Siguiendo el procedimiento anterior, y conociendo la Keq= 1023.5
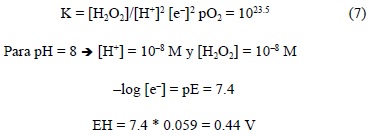
Este valor de potencial es más cercano al encontrado en aguas naturales. La concentración del H2O2 corresponde a los valores medios reportados en aguas naturales (Cooper y Zika, 1983; Petasne y Zika, 1997; Sarthou et al., 1997).
Metodología
Para las mediciones del potencial redox utilizamos inicialmente un grupo de cinco electrodos inorgánicos inertes: platino, gráfito, carbón vitrificado, oro e iridio. Estas mediciones se llevaron a cabo bajo condiciones controladas de laboratorio con la finalidad de obtener resultados redox reproducibles.
El diseño experimental consistió en realizar mediciones simultáneas de pH, O2 y EH en agua de mar sintética amortiguada. El pH se midió con un electrodo de combinación marca Orion Modelo 81-02. El oxígeno, con un electrodo tipo Clark marca Orion Modelo 97-08. Para las mediciones del potencial redox (EH) se utilizó como electrodo de referencia un calomel comercial (calibrado en el laboratorio). Los electrodos redox utilizados fueron de Pt (electrodo marca Beckman), grafito, carbón vitrificado, Au (alambre de Au con 99.99% de pureza y 0.25 mm de diámetro, Aldrich®) e Ir (alambre de Ir con 99.8% de pureza y 0.5 mm de diámetro, Johnson Matthey®). Los electrodos de Au, Ir y grafito fueron diseñados en nuestro laboratorio.
Las variaciones de oxígeno en la solución se efectuaron mediante burbujeo de gas nitrógeno (grado industrial con una trampa para gas oxígeno, Oxiclear®, modelo DGP-250-R2) para desplazar el oxígeno de la solución. De manera opuesta, se burbujeó aire cuando se deseó aumentar la concentración de oxígeno. Los cambios de pH se hicieron mediante la adición de HCl o NaOH, según el caso. La preparación del agua de mar sintética con una salinidad de 35 ups y una fuerza iónica de 0.7225; se preparó de acuerdo con Dickson (1993). Todas las sustancias utilizadas fueron grado reactivo. Las mediciones de los potenciales se obtuvieron con dos potenciómetros marca Orion 720A y los resultados se grabaron en una computadora personal a cada 30 segundos. Asimismo, se usó el programa de computación MINEQL+ (Schecher y McAvoy, 1998) para calcular el pE termodinámico en cada experimento.
Se efectuaron cuatro tipos de experimentos diseñados para estudiar el comportamiento y la respuesta de los distintos electrodos bajo condiciones comparables en el laboratorio:
(1) Se mantuvo constante el pH y se varió la concentración de oxígeno en la solución. Por otra parte, se realizaron otra serie de experimentos donde se mantuvo constante el oxígeno y se varió el pH mediante la adición controlada de HCl o NaOH, según fuera el caso.
(2) Se prepararon soluciones a diferentes concentraciones de pares redox en agua de mar sintética amortiguada a pH ~8.1 (Dickson, 1993) para conocer la respuesta comparada de los electrodos de Ir y Pt. Esta respuesta fué, además, comparada con la que predice el modelo.
(3) Se tomaron datos de pE en agua de mar, en la planicie lodosa de Bahía Falsa en San Quintín, durante un flujo y reflujo de marea, con los electrodos de Ir y Pt (de manera independiente). También se monitoreo el pE con un CTD acoplado a un electrodo de Pt, además de pH y O2.
(4) Finalmente, se analizaron muestras de agua de mar natural sin filtrar y filtrada através de un filtro de 1 µm y pasada por ultravioleta.
Las muestras fueron tomadas del laboratorio de acuacultura del Instituto de Investigaciones Oceanológicas de la Universidad Autónoma de Baja California (México).
Resultados y discusión
Los resultados obtenidos después de las mediciones redox en una solución de agua de mar sintética amortiguada (pH~8.1) con los diversos electrodos se muestran en la figura 1. Los cambios en la presión parcial de oxígeno deben reflejarse en el potencial redox dado que el pE es una función tanto de la presión parcial de oxígeno como del pH. Como se puede observar, las respuestas de los electrodos fueron muy diferentes. El electrodo de mejor respuesta potenciométrica, más cercana a lo que predice el modelo termodinámico, fue el de Ir.
Los electrodos de Au, carbón vitrificado y carbón grafito mostraron poca respuesta a los cambios de presión de oxígeno en la solución. Con base en los resultados obtenidos en esta parte experimental, se seleccionó el Ir como el mejor material para usarse como electrodo redox. Su respuesta electroquímica se compara con la del electrodo de Pt, dado que éste es el sensor redox más utilizado en la actualidad.
Es importante mencionar que el pE calculado se realizó con base en la hipótesis de que la pareja redox dominante en los sistemas marinos óxicos es la O2/H2O2, con una concentración del peróxido de hidrógeno de 10-8 M (Cooper y Zika, 1983; Petasne y Zika, 1997; Sarthou et al., 1997). Se seleccionó este valor de peróxido con base en resultados experimentales en los que se obtuvo un valor de EH de alrededor de 440 a 500 mV. De aquí, se calculó la concentración de peróxido que permitiera ese valor de EH. Este valor coincide con valores medidos en el medio natural (Dryssen y Wedborg, 1980; Cooper y Zika, 1983).
Se ha visto que el H2O2 es un intermediario clave en los procesos redox que involucran al oxígeno en procesos químicos y biológicos. Debido a su naturaleza reactiva y a su influencia en numerosos procesos químicos, el peróxido se ha sugerido como pareja redox controladora del pE en el medio ambiente marino (Petasne y Zika, 1997). Se puede decir que el sistema O2===>H2O2===>O2 muestra tiempos de vida media que va desde horas hasta días en el medio natural. Por lo tanto, se puede considerar a este sistema como un seudo-equilibrio con capacidad suficiente como para influenciar, en la presencia de metales como Mn(II), Fe(III), Pu(V) y As (III), entre otros, y dominar el sistema redox (Sato, 1960; Silver, 1991; Sarthou et al., 1997; Petasne y Zika, 1997).
En la figura 2a, se muestra la respuesta, pE, de los electrodos de Ir y Pt, ante los cambios de la concentración de O2 con respecto al tiempo manteniendo el pH constante. Se muestra también el comportamiento del pE que predice el modelo utilizando los cambios experimentales de O2 y pH. La respuesta de ambos electrodos ante los cambios de oxígeno es diferente. El electrodo de Ir muestra una tendencia a seguir más de cerca los cambios redox influenciados por los cambios de oxígeno. La principal desventaja observada del electrodo de Pt es que fue menos sensitivo a los cambios de presión de oxígeno. Estas diferencias se pueden interpretar como una respuesta electroquímica más rápida del electrodo de Ir que la del electrodo de Pt. En otras palabras, si se le permite al electrodo de Pt continuar en contacto con la solución, en un tiempo relativamente largo alcanzará el valor predicho termodinamicamente.

En la figura 2b se graficaron las respuestas del pE calculado contra el pE medido, para cada electrodo, utilizando los valores mostrados en la figura 2a. Las pendientes obtenidas en cada curva (modelo = 1, Ir = 0.88 y Pt =1.82) muestran que la respuesta electroquímica del electrodo de Ir se asemeja más al modelo termodinámico. Esta respuesta corresponde a una pendiente esperada con base en la ecuación de Nernst, en la que el intercambio de electrones sea de uno. En cambio, el electrodo de Pt presentó una pendiente superior a la esperada (respuesta super Nerstiana), que pudiera deberse a una cinética lenta del electrodo. Adicionalmente se puede mencionar que el electrodo de Ir presenta una mayor histéresis que la del de Pt.
En la figura 3a se observa la respuesta, pE, de los sensores Pt e Ir ante los cambios de pH con respecto al tiempo. Durante estas mediciones, la presión de oxígeno de la solución se mantuvo constante. En esta gráfica, el comportamiento de ambos electrodos a pHs básicos, responden de acuerdo a las predicciones de la termodinámica. Sin embargo, se debe recordar que el Pt responde como un sensor de pH. En la parte ácida, la respuesta del electrodo de Ir mostró ser más cercana a la predicha por el modelo.
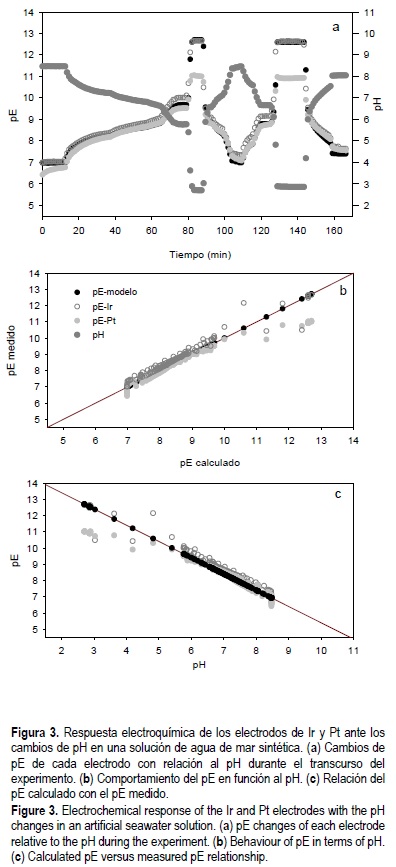
En la figura 3b se presentan los datos mostrados de pE medidos contra el pH de los datos mostrados en la figura 3a. En ella se reconfirma que la respuesta electroquímica del electrodo de Ir posee una pendiente que obedece a la ecuación de Nernst de -59 mV, en cambio, el electrodo de Pt mostró una pendiente de -53.1 mV en el mismo experimento. De nuevo, el Ir mostró un comportamiento más cercano al termodinámico.
En la figura 3c se muestra el comportamiento del pE medido contra el calculado, para los datos de la figura 3a. Es de nuevo evidente, en esta figura, que la respuesta del electrodo de Ir es mejor que la del de Pt.
Para comparar el comportamiento frente a pares redox en soluciones sencillas, se prepararon diferentes pares redox disueltos en una solución de agua de mar sintética amortiguada (pH ≅ 8.1). En ella, se midieron sus potenciales redox de manera independiente. Adicionalmente, se efectuaron mediciones de mezclas de estos pares como sistemas un poco más complejos.
En la tabla 1 se muestran los resultados de esta serie de experimentos. En ellos se observa que la respuesta de ambos electrodos es relativamente alejada de lo que predice el modelo, y sólo se acerca a lo predicho en aquellos casos en que el pE es cercano al que muestra el agua de mar sintética amortiguada (pE ≅ 6.8). Sin embargo, la poca diferencia entre ambos electrodos no es clara. Entre las posibles causas a las que pueda deberse este comportamiento se encuentran aquellas que mencionan Grente et al. (1992), entre ellas, que el sistema no posea suficiente capacidad redox para ser detectada por los sensores. Otra causa posible es la de que las mediciones se efectuaron en un tiempo relativamente corto y, posiblemente, los electrodos requirieran de mayor tiempo para alcanzar el equilibrio. Esto significaría que sus respuestas sean relativamente lentas.
En la tabla 2 se muestran los resultados obtenidos de la medición del pE en agua de mar natural y agua de mar sintética. En ella se observa una diferencia de aproximadamentre tres unidades de pE entre ambos electrodos. Las lecturas que se obtuvieron fueron muy estables durante las mediciones, lo que nos indica que el sistema se encontraba bien equilibrado y, por lo tanto, posee una alta capacidad redox.
Es importante hacer notar que el modelo utilizado para calcular el pE es un modelo sencillo de agua de mar sintética, que no toma en cuenta ni la presencia de la materia orgánica ni la de otros componentes importantes del sistema acuoso natural. Por esta razón, la comparación con el valor de pE calculado que se presenta en la tabla no permite decidir cuál de los dos electrodos se asemeja más al valor verdadero de pE.
Otra de las características que muestran los valores de la tabla es la de que ambos electrodos fueron relativamente insensibles a la presencia o ausencia de materia orgánica, ya que los valores obtenidos no fluctuaron mucho.
Si pensáramos en el orden decreciente de las reacciones de reducción del sistema marino natural y escribiéramos las correspondientes ecuaciones de pH-pE excluyendo el par redox O2/H2O, dado que hemos mostrado que este potencial (pE = 12) no se alcanza en la naturaleza, el siguiente par es N2/ NO3-, para el que se predice un pE mayor a 10 utilizando concentraciones normales para aguas locales. Un tercer par es el Mn(II)/Mn(IV), con un pE igualmente mayor a 10. Finalmente se pensaría en la pareja NO3-/NO2-, par redox que responde a un pE de 6.6 (Langmuir, 1997). Esto pudiera explicar la respuesta de los electrodos de Ir y Pt en un medio ambiente natural. La respuesta del Ir sugiere la presencia de H2O2 y NO3-.
En la figura 4 se muestran los resultados obtenidos en las mediciones del pE en la planicie lodosa de Bahía Falsa (San Quintín, Baja California). Se presentan los resultados independientes de los electrodos de Ir y Pt, junto con el electrodo redox (Pt) del CTD, todos ellos en función del tiempo.

Primeramente se puede observar que el electrodo de Pt del CTD no mostró mucha variación con respecto al tiempo, a pesar de los grandes cambios de oxígeno. En cambio, los electrodos independientes de Pt e Ir si mostraron una clara tendencia a responder a las fluctuaciones de oxígeno en el medio. Esto es, dado que la tendencia del medio fue a disminuir su contenido de oxígeno, los electrodos independientes respondieron midiendo pEs bajos. Cuando esta respuesta es comparada con la calculada utilizando un modelo sencillo de agua de mar, se observa que el Ir presenta una respuesta más cercana y más estable que la del Pt independiente.
Comparativamente, el Ir mostró una respuesta electroquímica relativamente más rápida que la del Pt.
Conclusiones
El iridio funciona como un sensor redox de rápida respuesta electroquímica, reproducible y confiable. Ante los cambios de oxígeno, el Pt mostró una respuesta electroquímica más lenta que el Ir. Ante cambios de pH, ambos electrodos funcionaron bién a pHs básicos, en cambio, a pHs ácidos la respuesta del Ir es más rápida que la mostrada por el Pt.
La pareja dominante en este sistema acuoso óxico, tanto en ambientes naturales como en los experimentos de laboratorio, es O2/H2O2, ya que las concentraciones de otros pares redox, en la solución, es tan baja que éstos no muestran la suficiente capacidad redox como para ser mostrada o identificada por los sensores redox.
Es evidente la necesidad de realizar mediciones continuas con este tipo de soluciones, así como estudiar más de cerca la cinética de estas reacciones.
Referencias
Baas-Becking, L.G.M., Kaplan, I.R. and Moore, D. (1960). Limits of the natural environment in terms of pH and oxidation-reduction potential. J. Geol., 68(3): 243-284. [ Links ]
Barcelona, M.J. and Holm, T.R. (1991). Oxidation-reduction Capacities of Aquifer Solids. Environ. Sci. Technol., 25(9): 1565-1572. [ Links ]
Bates, R.G. (1964). Determination of pH. Theory and Practice. John Wiley, New York. [ Links ]
Breck, W.G. (1972). Redox potentials by equilibration. J. Mar. Res., 30(1): 121-139. [ Links ]
Breck, W.G. (1974). Redox levels in the Sea. In: E.D.. Goldberg (ed.), The Sea. Vol. 5. Wiley-Interscience, New York, pp. 153-179. [ Links ]
Bricker, O.P. (1982). Redox potential: Its measurement and importance in water systems. Wat. Anal., Vol 1. [ Links ]
Champ, D.R., Gulens, J. and Jackson, R.E. (1979). Oxidation-reduction sequences in ground water flow systems. Can. J. Earth. Sci., 16: 12-23. [ Links ]
Chester, R. (1990). Marine Geochemistry. Unwin Hyman, London. [ Links ]
Cooper, L.H.N. (1937). Oxidation-reduction potential in sea water. J. Mar. Biol. Ass. UK, 22: 167-176. [ Links ]
Cooper, W.J. and Zika, R.G. (1983). Photochemical fomation of hydrogen peroxide in surface and ground waters exposed to sunlight. Science, 220: 711-712. [ Links ]
Dickson, A.G. (1993). pH buffer for sea water media based on the total hydrogen in concentration scale. Deep Sea Res., 40: 107-118. [ Links ]
Domenico, P.A. and Schwartz (1998). Physical and Chemical Hydrogeology. John-Wiley. [ Links ]
Dryssen, D. and Wedborg, M. (1980). Chemical speciation in estuarine waters. In: E. Olausson and I. Cato (eds.), Chemistry and Biogeochemistry of Estuaries. John Wiley. [ Links ]
Grente, I., Stumm, W., Laaksuharju, M., Nilsson, A.C. and Wikberg, P. (1992). Redox potential and redox reaction in deep groundwater systems. Chem. Geol., 98: 131-150. [ Links ]
Langmuir, D. (1997). Aqueous Environmental Geochemistry. Pretice- Hall, New Jersey. [ Links ]
Mansfield, C. (1925). Recent studies on reversible oxidation-reduction in organic systems. Chem. Rev., 2: 127-178. [ Links ]
Morel, F.M.M. and Hering, J.G. (1993). Principles and Applications of Aquatic Chemistry. Wiley-Interscience. [ Links ]
Morris, J.C. and Stumm, W. (1967). Redox equilibria and measurement of potentials in the aquatic environment. Advan. Chem. Ser., 67: 270-285. [ Links ]
Parsons, R. (1978). The kinetics of redox processes in aqueous solutions. Thalassia Jugoslavica, 14(1/2): 193-195. [ Links ]
Peiffer, S., Klemm, O., Pecher, K. and Hollerung, R. (1992). Redox measurements in aqueous solutions: A theorical approach to data interpretation, based on electrode kinetic. J. Contam. Hydrol., 10: 1-18. [ Links ]
Petasne, R.G. and Zika, R.G. (1997). Hydrogen peroxide lifetimes in south Florida coastal and offshore waters. Mar. Chem., 56: 215-225. [ Links ]
Pettine, M. and Millero, F.J. (1990). Chromium speciation in seawater: The probable role of hydrogen peroxide. Limnol. Oceanogr., 35(3): 730-736. [ Links ]
Pettine, M. (2000). Redox processes of chromium in sea water. In: A. Gianguzza, E. Pelizzatti and S. Sammartano (eds.), Chemical Processes in Marine Environments. Springer, New York, pp. 281-296. [ Links ]
Sarthou, G., Jeandel, C., Brisset, L., Amouroux, D., Besson, T. and Donard, O.F.X. (1997). Fe and H2O2 distributions in the upper water column in the Indian sector of the Southern Ocean. Earth Planet. Sci. Lett., 147(1-4): 83-92. [ Links ]
Sato, M. (1960). Oxidation of sulfide ore bodies. 1. Geochemical environments in terms of the Eh and pH. Econ. Geol., 55: 928-961. [ Links ]
Sato, M. and Mooney, H.M. (1960). The electrochemical mechanism of sulfide self-potentials. Geophysics, V. XXV (1): 226-249. [ Links ]
Schecher, W.D. and McAvoy, D.C. (1998). MINEQL+. A Chemical Equilibrium Modeling System. User's Manual. Environmental Research Software, Hallowell, Maine. [ Links ]
Sillén, L.G. (1952). Redox diagrams. J. Chem. Educ., 29: 600-608. [ Links ]
Sillén, L.G. (1967). Master variables and activity scales. Advan. Chem. Ser., 67: 45-55. [ Links ]
Silver, G.L. (1991). Environmental plutonium: What is the redox potential of seawater? J. Radioanal. Nucl. Chem. Lett., 155(3): 177-181. [ Links ]
Stumm, W. (1978). What is the pe of the sea? Thalassia Jugoslavica, 14(1/2): 197-208. [ Links ]
Teasdale, P.R., Minett, A.I., Dixon, K., Lewis, T.W. and Batley, G.E. (1998). Practical improvements for redox potential (EH) measurements and the application of a multiple-electrode redox probe (MERP) for characterising sediment in situ. Anal. Chim. Acta, 367: 201-213. [ Links ]
Thorstenson, D.C. (1984). The concept of electron activity and its relation to redox potencials in aqueous geochemical systems. US Geol. Surv. Open-File Rep. 84-072. [ Links ]
Truesdell, A.H. (1968). The adventage of using pE rather than Eh in redox equilibrium calculations. J. Geol. Educ., 16:17-20. [ Links ]
Vershinin, A.V. and Rozanov, A.G. (1983). The platinum electrode as an indicator of redox environment in marine sediments. Mar. Chem., 14: 1-15. [ Links ]
Whitfield, M. (1969). Eh as an operational parameter in estuarine studies. Limnol. Oceanogr., 14: 547-558. [ Links ]
Whitfield, M. (1974). Thermodynamic limitations on the use of the platinum electrode in Eh measurements. Limnol. Oceanogr., 19: 857-865. [ Links ]
Williams, P.A. (1990). Oxide Zone Geochemistry. Ellis-Horwood. Zobell, C.E. (1946). Studies on redox potential of marine sediments. Am. Assoc. Pretrol. Geol., 30(4): 447-513. [ Links ]

