











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de Joubert (SJ) es un grupo de trastornos clínica y genéticamente heterogéneos. Fue descrito por Marie Joubert en 1969. Tiene una prevalencia entre 1/80,000 y 1/100,000 nacidos vivos; pero en población judía Askenazi la prevalencia se incrementa hasta 1/34,000-1/40,000.1-3
En el SJ clásico, las manifestaciones clínicas pueden observarse desde los primeros meses de vida, con hipotonía, movimientos oculares anormales como nistagmos, estrabismo o apraxia oculomotora; además de alteraciones respiratorias (taquipnea o episodios de apnea), discapacidad intelectual y ataxia. En imágenes de resonancia magnética (IRM) cerebrales, el signo del diente molar (SDM) es el hallazgo característico, y consiste en la coexistencia de hipoplasia o displasia del vermis cerebeloso y de las estructuras pontinas y medulares, así como la falta de decusación de los pedúnculos cerebelosos superiores y de los tractos piramidales.4,5
El SJ puede combinarse con un amplio espectro de defectos orgánicos, por lo cual se conoce como “síndrome de Joubert y trastornos relacionados” (JSRD, por sus siglas en inglés). En el JSRD se identifican ocho fenotipos o subtipos que se describen en la Tabla 1.6,7
Tabla 1: Características clínicas de los ocho subtipos síndrome de Joubert*.
| Subtipos clínicos | Manifestaciones clínicas | Mutación en gen |
|---|---|---|
| SJ clásico | Hipotonía, nistagmos, estrabismo o apraxia oculomotora, taquipnea o episodios de apnea, retraso en el desarrollo neurológico y ataxia | TMEM67 |
| SJ con enfermedad de retina | Retinopatía pigmentaria o distrofia de la retina, amaurosis congénita de Leber’s. Retraso en el desarrollo neurológico | AHI1 |
| ARL3 | ||
| CEP290 | ||
| SJ con defectos renales | Nefronoptisis, quistes renales | CEP290 |
| RPGRIP1L | ||
| ZNF423 | ||
| TMEM231 | ||
| TMEM237 | ||
| SJ con enfermedad ocular-renal | Retraso en el desarrollo neurológico. Nefronoptisis, quistes renales. Retinopatía pigmentaria o distrofia de la retina. Retraso en el desarrollo neurológico | CEP290 |
| SJ con enfermedad hepática | Manifestaciones clínicas de SJ clásico. Fibrosis hepática congénita e hipertensión portal secundaria. Colobomas coriorretinianos o del nervio óptico y nefronoptisis | TMEM67 |
| SJ con defectos orofaciodigitales o tipo VI | Retraso en el desarrollo neurológico. Labio y paladar hendido, lengua lobulada, hamartomas de las encías o de la lengua y frenulas. Hipertelorismo y micrognatia o mandíbula. Polidactilia de manos y pies | TMEM216 |
| SJ con defecto acrocalloso | Disgenesia de cuerpo calloso, síndrome acrocalloso con polidactilia e hidrocefalia | KIF7 |
| SJ con distrofia torácica asfixiante de Jeune | Talla baja, tórax estrecho, acortamiento rizomélico de extremidades, braquidactilia y polidactilia | TMEM216 |
* En los ocho siempre está presente el signo del diente molar.
Se ha establecido una correlación entre el SJ y las variantes en el gen TMEM67 (proteína transmembrana-67). Las variaciones de la secuencia bialélica de TMEM67 causan una amplia gama de manifestaciones clínicas con compromiso multiorgánico como SJ (MIM 610688), síndrome de Meckel-Gruber (MIM 607361), síndrome COACH (MIM 216360) (hipoplasia de vermis cerebeloso, oligofrenia, ataxia, coloboma y fibrosis hepática), síndrome RHYNS (MIM 613550) (rinitis pigmentosa, hipopituitarismo, nefronoptisis, displasia esquelética); además, los pacientes con SJ con mutaciones en TMEM67 tienen mayor riesgo de desarrollar enfermedad hepática.8
El objetivo de este estudio es presentar un caso de SJ clásico con dos variantes de TMEM67.
Presentación del caso
Niño de tres años, sin antecedentes familiares de enfermedades genéticas; padres no consanguíneos. Es producto de la gestación cuatro; nació por parto eutócico a las 40 semanas de edad, sin antecedente de hipoxia neonatal, peso 3,020 g y talla de 48 cm. A los 57 días de edad presentó tosferina (cultivo positivo para Bordetella pertussis), síndrome de dificultad respiratoria aguda, y paro cardiorrespiratorio por varios minutos, recibió ventilación mecánica por nueve días en la unidad de cuidados intensivos pediátricos, permaneciendo 32 días hospitalizado.
Aproximadamente a los tres meses de edad, los padres observaron movimientos oculares anormales, ausencia de sostén cefálico, hipotonicidad generalizada, sonrisa social débil y falta de persecución visual.
A los cuatro meses fue evaluado por oftalmología por los movimientos anormales de los ojos, prescribiendo lentes monofocales (Figura 1). Sin embargo, a los ocho meses de edad nuevamente los padres lo llevan por inatención visual, estrabismo convergente con predominio del ojo izquierdo (con o sin lentes), y nistagmo vertical hacia abajo (Figura 2). En la exploración oftalmológica se encontró agudeza visual en ambos ojos de gradiente 0.25 ciclos por centímetro (CPCM), equivalente a 20/2000 Snellen, lo cual le permitía fijar, mantener y seguir objetos por segundos. Además, se describió endotropía de Hirschberg de 30º en ojo izquierdo con escasa alternancia, con y sin corrección óptica. Cuando le colocaron lentes monofocales con la siguiente graduación: + 6.00-0.00 × 180 en el ojo derecho y + 6.00-0.00 × 0 en el izquierdo, presentó un estrabismo convergente (Figura 2). También se detectó estrabismo vertical (hipotropía) ipsilateral con movimientos verticales involuntarios de gran frecuencia y amplitud, versiones y ducciones sin restricción. La anatomía de la cámara anterior se observó íntegra.


Figura 2: Estrabismo convergente por test de Hirschberg de ojo izquierdo sin lentes. No hay rasgos faciales dismórficos.
Se realizó estudio de fondo de ojo bajo midriasis medicamentosa (tropicamida y fenilefrina), además de oftalmoscopía indirecta. Se observó que en ambos ojos las papilas eren de forma, tamaño y coloración normales, con excavaciones de 0.3 × 0.3, simétricas; área macular normal, sin alteraciones pigmentarias en polo posterior ni en media periferia (Figura 3). Además, la refracción bajo ciclopejía (con ciclopentolato) fue en ojo derecho (OD) de + 7.25 = -1.50 × 160, y en ojo izquierdo (OI) de + 7.00 = -1.50 × 0. Asimismo, al medir las amplitudes acomodativas (retinoscopía dinámica por método de estimulación monocular (MEM) se detectó hipoacomodación, estableciendo el diagnóstico de endotropía acomodativa refractiva con hipoacomodación central, ambliopía estrábica y nistagmo vertical hacia abajo. Por lo anterior, se prescribieron lentes hipercorregidos (Figura 4), con graduaciones en el OD: + 10.25-1.50 × 160 y OI: + 10.00-1.50 × 0. Posteriormente se dio vigilancia cada dos meses para evaluar el desarrollo visual, se observó mejoría tanto de la alineación ocular como de la atención visual, logrando alternancia y disminución de la dominancia ocular.


A los 18 meses de edad se observó una franca mejoría en su atención visual, presentó una agudeza visual de gradiente 8.0 CPCM (20/80 cartillas de Snellen) en ambos ojos. En movilidad ocular se mostró una ortoposición (Figura 4), con persistencia del nistagmo vertical hacia abajo, pero con mayor control. Además, se detectó asimetría en el nistagmo optocinético, pero la presión intraocular (medido con tonómetro de iCare) del OD fue de 11 mmHg y en el OI de 12 mmHg.
Por otro lado, también a los ocho meses de edad se realizó RM de encéfalo, la cual mostró pedúnculos cerebelosos superiores alargados con configuración del SDM (Figura 5). De esta forma, al integrar las características clínicas del paciente, con hipotonía, nistagmo, retraso en el desarrollo psicomotor y la imagen SDM se llegó a la conclusión de que se trataba de un SJ clásico.
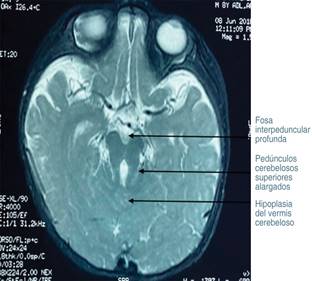
Figura 5: Corte coronal de resonancia magnética cerebral donde se observan pedúnculos cerebelosos superiores alargados con configuración del “signo del diente molar”.
Para identificar la posible mutación, a la edad de tres años se realizó análisis molecular mediante secuenciación de siguiente generación, empleando el DNeasy Blood & Tissue Kit de QIAGEN. En preparación de la librería del ADN genómico se empleó el kit xGen Exome Research Panel v1.0. La secuenciación de todo el exoma fue con la plataforma Illumina NextSeq 550 System en una configuración 2X 159bp-extremo emparejado. Por último, se realizó análisis bioinformático. Con todo lo anterior, fue posible encontrar dos variantes en el gen TMEM67, c.1322G>T (p.Arg441Leu) del exón 13 y c.431T>G (p.Leu144Trp) del exón. De acuerdo con los tipos de software predictivos LTR, SIFT, PolyPhen y Mutation Taster se determinó que ambas variantes son probablemente patogénicas. Pero no se pudo determinar si estas variantes se encuentran en el mismo (cis) o en cromosomas opuestos (trans). Tampoco fue posible realizar el estudio genético a los padres, pero dadas las características clínicas del paciente, inferimos que ambas variantes se encuentran en estado de heterocigocidad compuesta.
Es necesario aclarar que durante todo el tiempo descrito en el presente reporte, el paciente ha recibido terapia física y de rehabilitación en virtud del retraso para la deambulación, el habla y el lenguaje; además, la función renal y hepática se ha mantenido dentro de lo normal.
Discusión
Las ciliopatías son un grupo de trastornos genéticos causados por la formación o función anormal de los cilios primarios celulares en diferentes órganos. Hasta la fecha, se sabe que más de 35 genes causan SJ, con herencia autosómica recesiva, mientras que solamente se ha identificado un gen ligado al cromosoma X; en conjunto, estos genes representan entre 62 y 94% de los casos de SJ.7-9
Las variantes en el gen TMEM67 representan aproximadamente 80% de los pacientes con SJ con afectación hepática por fibrosis hepática congénita; ésta es la característica principal en este subtipo, y los pacientes tienen alto riesgo de desarrollar hipertensión portal en la segunda o tercera década de la vida.10 Brooks y colaboradores evaluaron las alteraciones oculares en 99 pacientes con SJ, y en 95.9% se identificó la causa molecular: 22.2% mostraron una variante en el gen TMEM67, de los cuales 50% presentaron enfermedad renal, 95% enfermedad hepática, 73% apraxia oculomotora, 59% nistagmo, 87% estrabismo, y ptosis en 36%.11 A diferencia de ese reporte, nuestro paciente hasta la edad de tres años no ha presentado daño hepático o renal.
El gen TMEM67 ha sido mapeado a 8q22.1, tiene 29 exones y codifica para una proteína de 995 aminoácidos denominada meckelina.12 En estudios de hibridación in situ en embriones humanos se ha encontrado expresada la proteína meckelina en riñón, hígado, retina, romboencéfalo y línea media cerebral. Por inmunohistoquímica, en riñones fetales se ha encontrado expresión moderada de meckelina en el epitelio del túbulo renal proximal, pero no en el glomérulo. En hígado, estas proteínas también se expresan en el epitelio biliar de los conductos biliares, pero no en los hepatocitos.13 Estudios en modelos de ratón y en células de pacientes con ciliopatía semejante a MKS (Meckel-Gruber syndrome) y SJ sugieren que la proteína meckelina forma un complejo funcional con la proteína filamina A, que se interrumpe en pacientes con MKS y causa defectos en la migración neuronal y señalización de Wnt. Se sabe que la filamina A tiene un papel crucial en los procesos normales de ciliogénesis y posicionamiento basal del cuerpo. Simultáneamente con estos procesos, el eje de señalización de meckelina-filamina A puede ser un regulador clave para mantener niveles normales y correctos de señalización de Wnt.14,15 Wnt es una vía que regula aspectos del destino, migración y polaridad celular, así como del patrón neural y la organogénesis durante el desarrollo embrionario.15
Nuestro paciente presenta dos variantes probablemente patogénicas en el gen TMEM67, una es c.1322G>T (p.Arg441Leu) del exón 13, misma que ya ha sido descrita,10 y la otra c.431T>G (p.Leu144Trp) aún no descrita en la literatura ni en la base de datos ClinVar; sin embargo, por los tipos de software predictivos LTR, SIFT, PolyPhen y Mutation Tester, la variante es probablemente patogénica.
Dado que no se observa hasta el momento daño hepático ni renal, de acuerdo a pruebas bioquímicas en sangre y ecografía renal y hepáticas, sólo es posible clasificarla en el subtipo SJ puro o clásico; sin embargo, dada las proporciones arriba comentadas, que describen variantes en el gen TMEM67, sumado a la heterogeneidad clínica y genética del SJ nos hace suponer la alta probabilidad de que la enfermedad hepática, principalmente, se encuentre dentro del curso natural de la enfermedad en este paciente, por lo que de ocurrir a una edad mayor, correspondería al subtipo SJ con enfermedad hepática.
Es muy importante la asesoría genética ante un caso de SJ, por el riesgo de recurrencia de 25% en las formas hereditarias autosómica recesivas. La detección prenatal de SJ es posible mediante ecografía fetal a partir de la semana 11 a 12 de gestación, y de evaluación de la anatomía cerebelosa fetal con RM entre las 20 y 22 semanas de gestación.
El niño con nistagmo, déficit intelectual y ataxia debe ser sometido a un examen exhaustivo ocular y de neuroimagen. El tratamiento en el niño con SJ debe estar dirigido a estrategias de rehabilitación adecuadas, tratar el déficit motor, de lenguaje y visual. El pronóstico en el paciente depende del tipo JSRD y del número de lesiones orgánicas.16

