











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink1. Introduction
Spintronics is one of the key areas for the future-generation nanotechnology devices to increase their memory and reduce their power consumption. After the discovery of the giant magnetoresistance (GMR) [1,2] effect, the half-metallic (HM) materials are becoming the most potential candidates for spintronic applications [3].
This class of materials show semiconducting or insulator behavior in one spin channel and metallic character in the other, which makes them suitable candidates for various spin-tronic applications, including the development of magnetic sensors (MS), non-volatile magnetic random access memories (NV-MRAM), spin valves (SVs) and spin-injector materials (SIMs) [4-7].
Half-metallic behavior has been reported to be exhibited in a wide range of materials such as zinc blende and wurtzite structured compounds [8-10], Heusler alloys [11-14] and per-ovskites [15,16].
In particular interest, Heusler alloys have attracted more both theoretical and experimental researchers to investigate over the last decades because of their many specific properties includingmagnetic shape memory [17,18], spin gap-less semiconductor [19-21], thermoelectric [22-24], superconductivity [25], and half-metalicity behavior [26,27].
The two well-known classes of Heusler alloys are half-Heusler and full-Heusler alloys. The former have a general chemical formula XYZ, and crystallize into C1b structure with F 43m space group, where X and Y represent transition metals, and Z stands for an sp-valent element while the latter have X2YZ stoichiometry, and crystallize into L21 structure with Fm3m space group [28].
A new class of half-Heusler alloys (HHAs) without transitionmetal elements called d0 (HHAs) have recently been-reported where the magnetism comes mainly from the anion p electrons. The d0 (HHAs) compounds seem to be more appropriate than others because they could display high Curie temperatures, low magnetic moments and similar crystal structures with semiconductors [29]. Numerous studies have been carried out on such half-Heusler based alloys. In a recently published work, Rozale et al. [30,31] confirmed the half metallicity behavior in half-Heusler compounds XRbSr (X = C, Si, and Ge) and XCsSr (X = C, Si, Ge, and Sn). In the same context, Benabboun et al.[32] have explored the magnetic behavior of d0 XCaZ (X =Li, Na; Z=B, C) half-Heusler and confrmed that the magnetism comes from atomic p orbitals, which persist in the alloys of calcium due to its large atomic radii. In another study, Abada and Marbouh [33] used density functional theory based calculations to study the MgCaB alloy predicting half-metallicity and the nearly spingapless nature of the alloy when the lattice constants lie in the range of 7.32-8.14 Å.
Similarly, many other researchers have determined the half metallicity in different materials including half-Heusler alloys XYZ (X = Li, Na, K, and Rb; Y = Mg, Ca, Sr, and Ba; Z = B, Al, and Ga) [34], XRbSr (X = C, Si, and Ge) alloys [35], NaKZ (Z=N, P, As, and Sb) compounds [36], LiXGe (X =Ca, Sr, and Ba) alloys [37] and MNaCs (M= P, As) [38].
Using these data, we seek to further search for new d0 (HM) materials with low magnetic moments and high spin polarisation such as CsCaZ (Z= Ge, Sn andPb) half-Heusler compounds. In this paper, the main objective is to investigate the physical propertiesincluding HM character, structural, electronic, magnetic, elastic and thermodynamic properties of CsCaZ (Z= Ge, Sn and Pb) compounds for potential spintronic applications.
2. Computational details
The calculations were performed within the spin-polarized density functional theory (SP-DFT) [39,40]. We used the full potential linear augmented-plane waves (FP-LAPW) method implemented in the Wien2k package [41]. The exchange-correlation potential was treated using Perdew-Burke-Ernzerhof generalized gradient (GGA-PBE96) approximation [42].The partial wavesused inside the atomic spheres are expanded up to lmax = 10 with a matrix size RMT × Kmax equal to 8, where Kmax is the plane wave cutoff and RMT is the smallest of all atomic sphere radii. The muffin-tin sphere radii automatically set in the calculations are 2.00, 2.00 and 1.93 Bohr for Cs, Ca and Z atoms, respectively. The value of the largest vector value in charge density Fourier expansion Gmax is 12. The cutoff energy, which defines the separation of valence and core states, was chosen as -6.0 Ry. We impose a convergence criterion of 10 - 5a.u.in the total energy to improve accuracy in the spin-polarized calculations.
3. Results and discussions
3.1. Structural stability
The half Heusler compounds (XYZ) are ternary intermetallic alloys. They crystallize in the face-centered cubic (fcc) C1b structure with the space group F 43 m (no. 216). For symmetry reasons, only three phases α, β, and γ are displayed in Table I.
Table I Inequivalent site occupancies within the C1b-type structure for CsCaZ (Z= Ge, Sn and Pb).
| Type/Atom | Cs | Ca | Z |
|---|---|---|---|
| α - Type | (0.25, 0.25, 0.25) | (0.75, 0.75, 0.75) | (0, 0, 0) |
| β - Type | (0, 0, 0) | (0.75, 0.75, 0.75) | (0.25, 0.25, 0.25) |
| γ - Type | (0.5,0.5,0.5) | (0.75, 0.75, 0.75) | (0, 0, 0) |
With the purpose of determining the stable ground state structure of CsCaZ, we implemented the volume optimizations for three types α, β, and γ only in the ferromagnetic (FM) phase, which is the most favorable state compared to the non-magnetic (NM) and the anti-ferromagnetic (AFM) phases. The unit cell of CsCaZ (Z = Ge, Sn and Pb) Heusler alloys in α - Type phase is shown in Fig. 1.
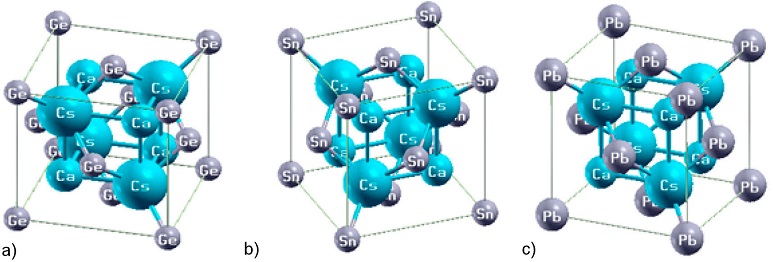
The results of the calculated total energies versus volume in different types of the materials investigated are represented in Fig. 2. From this figure, it can be seen that all compounds show the lowest total energy in α - Type. The optimized equilibrium parameters, including the lattice parameter (α), (α), the bulk modulus (β), and its first derivative (β) for the three compounds, were determined by the empirical Murnaghan's equation [43] and shown in Table II.
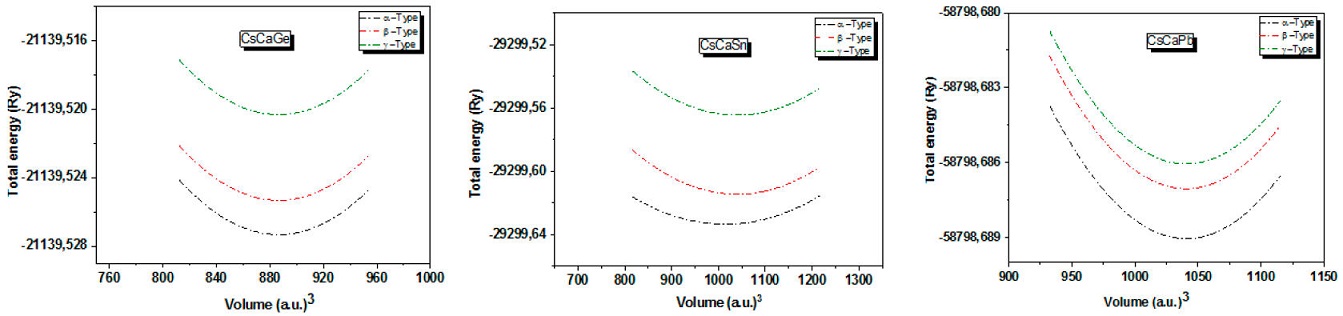
Figure 2 Total energy as a function of unit cell volume for the three types of ternary Heusler alloys CsCaZ (Z= Ge, Sn and Pb) using the GGA-PBE approximation.
Table II The calculated equilibrium lattice parameter (a), bulk modulus (B), derivative of bulk modulus (B'), the cohesive energy (Ec),the formation energy (Ef ) and the ground state energy (E0) of CsCaZ (Z = Ge), Sn and Pb.
| Compound | α (A0) | B (GPa) | B’ | EC (eV) | Ef (eV) | E0 (Ry) |
|---|---|---|---|---|---|---|
| CsCaGe | 8.00 | 16.44 | 4.85 | -1.24 | -1.75 | -21139.5274 |
| CsCaSn | 8.41 | 15.06 | 4.10 | -2.85 | -2.24 | -29299.6334 |
| CsCaPb | 8.52 | 13.94 | 3.75 | -4.97 | -4.18 | -58798.6891 |
The equilibrium lattice constants are found to be 8, 8.41 and 8.52 Å for CsCaGe, CsCaSn, and CsCaPb, respectively. This result can be explained by considering the atomic radii of Ge, Sn and Pb whuch are respectively 1.25, 1.45 and 1.54 A0. That is, the lattice constant increases with the atomic size of the Z element in CsCaZ compounds. However, the B value decreases in the following sequence: B (CsCaGe) > B (CsCaSn) > B (CsCaPb), conversely to (a) and the unit cell volume (V), which follow the relationship (B ~ V-1) [44].
To our knowledge, no experimental or theoretical data are available for comparison, so the present results will be useful for future research.
We also calculated the formation energy (Ef), which is the change in energy when a material is formed from its constituent elements in their bulk states. Ef per formula unit of CsCaZ (Z= Ge, Sn and Pb) compounds is given by the relation:
where,
We further calculated the cohesion energy Ec. This parameter represents the energy required to break the atoms of the solid into isolated atomic species.
The cohesive energy per formula unit of CsCaZ (Z= Ge, Sn and Pb) is given by the following formula:
where
According to Table II, the obtained values of Ec for all compounds are negative, which implies that the HHAs studied are expected to be stable and synthesized experimentally due to their high bonds energies [46].
3.2. Elastic properties
In order to confirm the mechanical stability of these materials, it is necessary to calculate their elastic constants (Cij). These quantities can be used to provide the critical information regarding the mechanical stability of a solid structure against the arbitrary deformation such as propagation of elastic waves in normal mode, specific heat and chemical bonds [47].
The elastic stability criteria for a cubic crystal [48] was examinated by using three independent elastic constants namely C11, C12, and C44.
The calculated elastic constants of the half-Heusler alloys CsCaZ (Z= Ge, Sn and Pb) in the α - Type structure are listed in Table III. It is observed that these values verify the above stability conditions,which is indicative of the mechanical stability of all compounds in this structure.
Table III The calculated elastic constants Cij (GPa), bulk modulus B (GPa), shear modulus G (GPa), Cauchy's pressure CP (GPa), Young's modulus E (GPa), Pugh's ratio B/G, anisotropy factor A and Poisson's ratio v for CsCaZ (Z= Ge, Sn and Pb).
| Compound | C11 | C12 | C44 | B | G | CP | E | B/G | A | v |
|---|---|---|---|---|---|---|---|---|---|---|
| CsCaGe | 31.27 | 9.35 | 2.32 | 16.56 | 4.58 | 7.03 | 12.58 | 3.62 | 0.21 | 0.39 |
| CsCaSn | 27.10 | 9.23 | 2.43 | 15.22 | 4.23 | 6.80 | 11.61 | 3.60 | 0.27 | 0.37 |
| CsCaPb | 24.15 | 8.95 | 2.56 | 14.01 | 4.03 | 6.39 | 11.03 | 3.48 | 0.34 | 0.36 |
Other important elastic moduli can be calculated applying the Voigt-Reuss-Hill (VRH) approximation [49]:
where, B is the bulk modulus, GV the Voigt's shear modulus, GR the Reuss's shear modulus, G the shear modulus, CP the Cauchy's pressure, v the Possion's ratio, A the anisotropy factor and E Young's modulus.
As shown in Table III, the obtained bulk modulus (B) values of 16.56, 15.22 and 14.01 GPa for CsCaGe, CsCaSn and CsCaPb, respectively, are in close agreement with the calculated values determined by fitting the total energy as a function of unit cell volume.
Since the calculated values of the Pugh's ratio (B/G) are above 1.75, the materials studied tend to be ductile [50].
The calculation of Cauchy pressure (CP) will determine the type of atomic bonding, with a negative value for directional covalent bonding and a positive value for non-directional metallic bonding.
Based on the Frantsevich rule[51],if the poisson's ratio v > 1/3, the material is considered ductile, otherwise it will be brittle.As shown in Table III, all the computed Poisson's ratios (v) are above 0.33, signifying that the Half Heusler Alloys CsCaZ (Z= Ge, Sn and Pb)are ductile in nature.
Furthermore, the higher the value of Young's modulus (E), the stiffer the compound will be and in this case, CsCaGe is the stiffest.
The Zener anisotropy factor (A) is equal to 1 for isotropic crystal and deviation from unity indicates an anisotropic crystal.It is observed that the calculated values of (A) are equal to 0.21, 0.27 and 0.34 for CsCaGe, CsCaSn and CsCaPb, respectively, which confirms the mechanical anisotropy of these compounds.
3.3.1. Band structure
In this part, wemainly focus on the spin-polarized electronic band structure of CsCaZ (Z= Ge, Sn and Pb) compounds along the high-symmetric direction of the Brillouin zone (BZ), in which the Fermi energy levels (EF) were set to zero (Figs. 3-5).For all compound, the majority spin channel (spin up) is foundto exhibit a semiconducting behavior, whereas, the spin minority channel (spin dn) shows a metallic character,resulting in stable half-metallic ferromagnetic behavior.
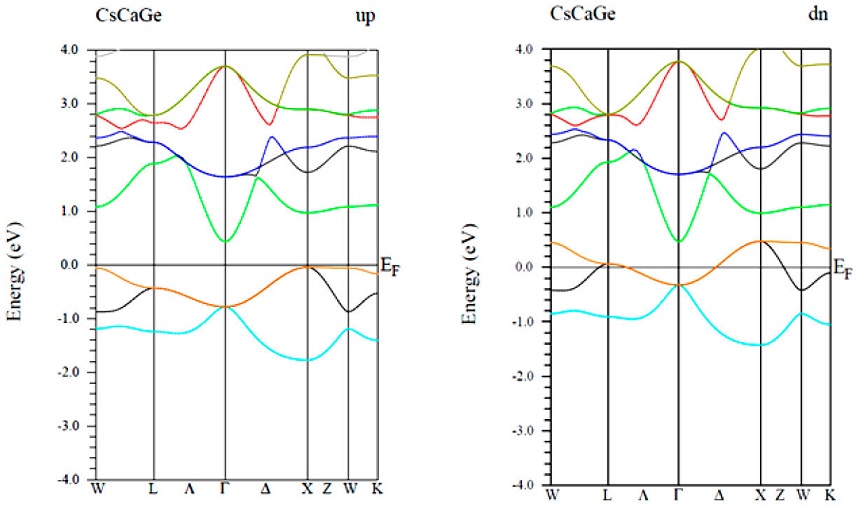
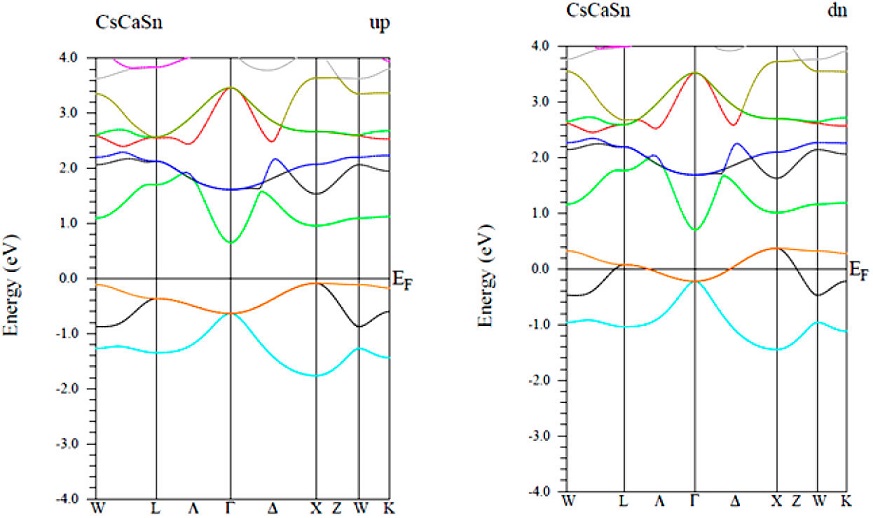
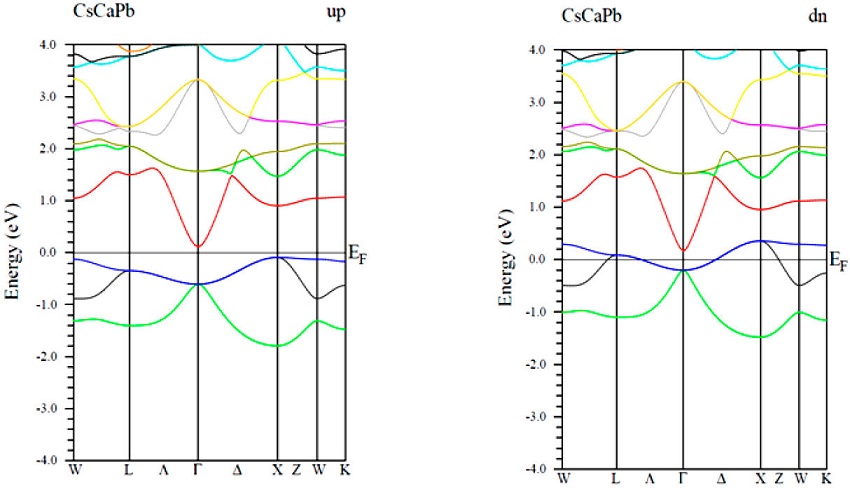
In the majority-spin channel, the indirect band gaps (IBG) at around EF along the Γ - X symmetry are of 0.42, 0.74 and 0.17 eV for CsCaGe, CsCaSn and CsCaPb respectively. This results leads to 100% spin polarization at EF for all material.So far, noexperimental measurements have been carried out for the band structure of these compounds to compare with. However, for other similar cubic ternary half-Heusler compounds, the size of their band gaps has been the oretecally established at 0.70 eV for CsBaC [52] and at 1.73, 1.61 and 1.23 eV for RbSrC, RbSrSi and RbSrGe, respectively [53].
3.3.2. Density of states (DOS)
To further elucidate the nature of the electronic band structure, we calculated the total (TDOS) and partial densities of states (PDOS) for both spin-up and spin-down states of CsCaZ (Z= Ge, Sn and Pb) alloys as depicted in Fig. 6. From the (TDOS), in spin-down channel, a peak crosses the Fermi level revealing the conductor behavior for both materials. However, in spin-up one, a semiconductor character is observed, confrming the half-metallicity of both materials already seen in band structures. From the (PDOS) in spin-down direction, it can be noted that 4p states of (Ge/Sn/Pb) are responsible for the metallicity of the three compounds with a week contribution of s-Cs and s-Ca states. In spin-up one, the lowest part of the valence band is mainly formed by the s-Cs and s-Ca states and the upper one comes exclusively from p-Z orbitals. In spin up configuration, a small energy gap is found where the p-Z state has made a major contribution in the valence band whereas the conduction band in both channels is dominated by s-Cs and s-Ca states. We can deduce that around the Fermi level, the p-orbitals of the main group element (Ge/Sn/Pb) are the major contributors to the density of states. As a result, these atoms will play a pivotal role in the transport properties of CsCaZ (Z= Ge, Sn and Pb) HH alloys.
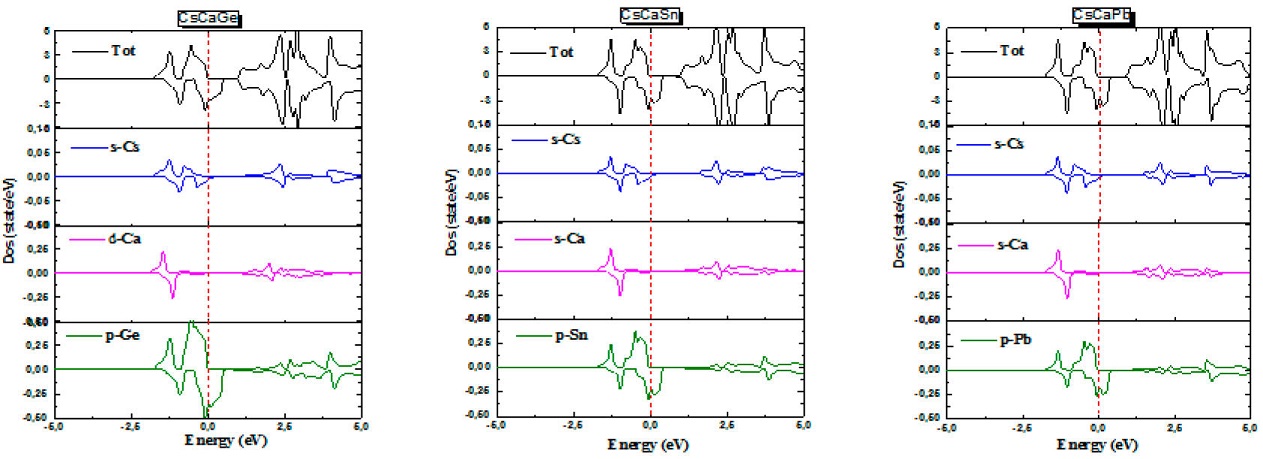
3.4. Magnetic properties
The calculated local and total magnetic moments in interstitial region for the studied half-Heusler CsCaZ (Z= Ge, Sn and Pb) compounds are presented in Table IV.
Table IV The calculated interstitial, partial and total magnetic moments per formula unit and energy gaps Eg (eV) of the CsCaZ (Z = Ge, Sn and Pb) compounds.
| Compound | MInt (μB) |
MCs (μB) |
MCa (μB) |
MZ (μB) |
Mtot (μB) |
Eg (eV) |
Band gap |
Classification |
|---|---|---|---|---|---|---|---|---|
| CsCaGe | 0.54 | 0.02 | 0.06 | 0.38 | 1.00 | 0.42 | Indirect | HMF |
| CsCaSn | 0.62 | 0.03 | 0.06 | 0.29 | 1.00 | 0.74 | Indirect | HMF |
| CsCaPb | 0.64 | 0.04 | 0.05 | 0.27 | 1.00 | 0.17 | Indirect | HMF |
The integer values obtained for the total magnetic moment (1μB), using the GGA approach, confirm a half-metallic behavior of these compound according to Slater-Pauling rule:
where Mtot is the total magnetic moment, and Ztot, the valence electron number. The value of Zt for CsCaZ (Z= Ge, Sn and Pb) is 7. So, seven valence electrons contribute to bonds and magnetism (Cs:6s1-; Ca: 4s2-; Z: ns2, np2). According to Eq. (9), the Mtot obtained is equal to 1μB for CsCaZ (Z = Ge, Sn and Pb), which is in good agreement with the calculated results of Table IV. Moreover, the calculated Mtot is in perfect accordance with available theoretical work performed on cubic ternary half-Heusler compounds LiBaX (X = Si, Ge), and GeNaZ (Z = Ca, Sr, and Ba) ternary half-Heusler alloys where their Mtot is equal to 1μB [54,55]. It's also seen from Table IV that the total magnetic moment is mainly relative to the main group element (Ge/Sn/Pb) and to the interstitial region.
3.4.1. Lattice parameter impact on half-metallicity
During the manufacturing of spintronic devices, stress can develop during the process of epitaxial growth due to lattice mismatches. For this reason, one of the most important characteristics that must be studied is the retention of the half metallic nature of our materials with variations in the lattice parameter.Furthermore, knowing that the integer magnetic moment is a requirement for the half-metallicity, we have studied the total magnetic moment change as function of lattice constant (Fig. 7).
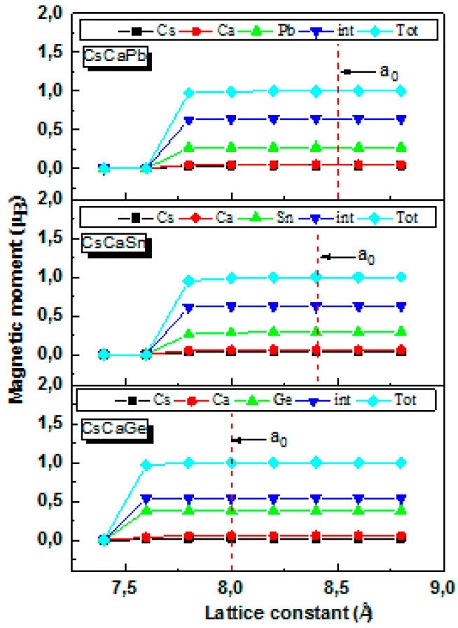
Figure 7 Calculated total and atomic spin magnetic moments as a function of lattice constant of CsCaZ (Z = Ge, Sn and Pb) Heusler alloys.
It can be seen that the total magnetic moment of 1μB, which obeys the Slater-Pauling rule, is well maintained in a wide range of lattice parameter values 7.6-8.8 Å for CsCaGe and 7.8-8.8 Å for CsCaSn/CsCaPb. Furthermore, for lattice contants beyond the above-mentioned range, the disappearance of the half-metallicity is not abrupt. The ranges in which the CsCaZ alloys agree with the Slater-Pauling rule are considerably wide, especially for the CsCaGe alloy. It also indicates that as the lattice constants increase, the atomic magnetic moments values remain stable in the interstitial, Cs, Ca and Z atoms, showing that the lattice contants have no effect on the magnetic properties of the compounds studied. As a result, one can say the half-metallicity of CsCaZ (Z= Ge, Sn and Pb) compounds is preserved with respect to lattice contants, which makes them potential candidates for spintronics applications as thin films.
3.5. Thermodynamic properties
In order to evaluate the specific behaviors of CsCaZ (Z= Ge, Sn and Pb) compounds in response to temperature and pressure, we have applied the quasi-harmonic Debye model (QHDM) as implemented in Gibbs program [56].The variation of the bulk modulus (B), Debye temperature (θD),heat capacityat constant volume (CV), thermal expansion (α) and entropy (S) are calculated under pressure range from 0 to 20 GPa and temperature range from 0 to 1200 K.
The non-equilibrium Gibbs free energy can be stated as:
where E(V) is the total energy per unit cell, P the pressure, V the unit cell volume, θD, the Debye temperature and Avib, the vibrational Helmholtz free energy term which will be expressed as [57],
where n is the number of atoms per formula unit, kB the Boltzmann constant and D(θD/T) the Debye integral.
The Debye temperature θD distinguishes the behavior of phonons between quantum mechanical and classical methods. It is calculated using the following classical relation [58],
where σ is Poisson's ratio, M the molecular mass per unit cell and BS the adiabatic bulk modulus.
f(a) is expressed as [59]:
The isothermal bulk modulus BT is given by:
The heat capacity Cv, the thermal expansion coefficient α and the entropy Sare calculated as follows [60],
Bulk modulus as a function of temperature for different pressure valuses for all compounds is depicted in Fig. 8.
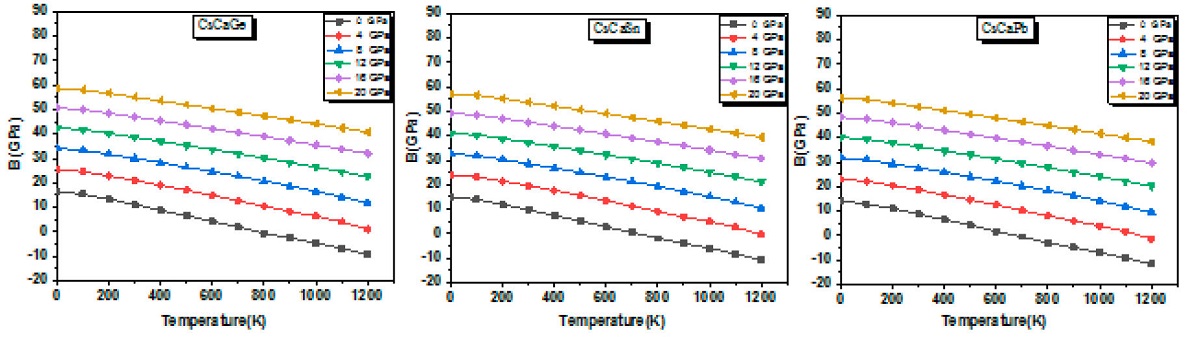
First, at constant temperature, the bulk modulus exhibits a near linear increasing variation with pressure. Next, negligible change of this parameter is observed in the temperature range from 0 to 100 K. However, after 100 K, it shows a significant decreasing trend when the temperature increases. However, the bulk modulus decreases more slowly at higher pressure. Our results show that the hardness of CsCaZ (Z = Ge, Sn and Pb) compounds will be enhanced at high pressure, whereas increasing temperature will cause a contrary behavior. In addition, at 0 K and 0 GPa conditions, the obtained bulk modulus values for CsCaGe, CsCaSn and CsCaPb are 16.45, 15.05 and 14.05 GPa, respectively, which are in good agreement with the previously determined ones using the structural and elastic properties. Furthermore, at ambient temperature and zero pressure (see Table V), the high bulk modulus values of 11.19, 9.79 and 8.79 GPa corresponding to CsCaGe, CsCaSn and CsCaPb,respectively, confirm that CsCaGe presents a strong hardness and an important compressibility [61].
Table V Calculated thermodynamic parameters: bulk modulus (B in GPa); acoustic Debye temperature (ΘD in K); thermal expansion coefficient (α); specific heat (CV in J/mol-1K) and entropy (S in in J/mol-1) for CsCaZ (Z= Ge, Sn and Pb) half-Heusler compounds (All parameters calculated at zero pressure and 300 K).
| Compound | B | Θd | Cv | α | S |
|---|---|---|---|---|---|
| CsCaGe | 11.19 | 184,59 | 71.95 | 22.14 | 91.44 |
| CsCaSn | 9.79 | 172.59 | 72,60 | 13.65 | 89.34 |
| CsCaPb | 8.79 | 160.59 | 73 | 9.64 | 86.33 |
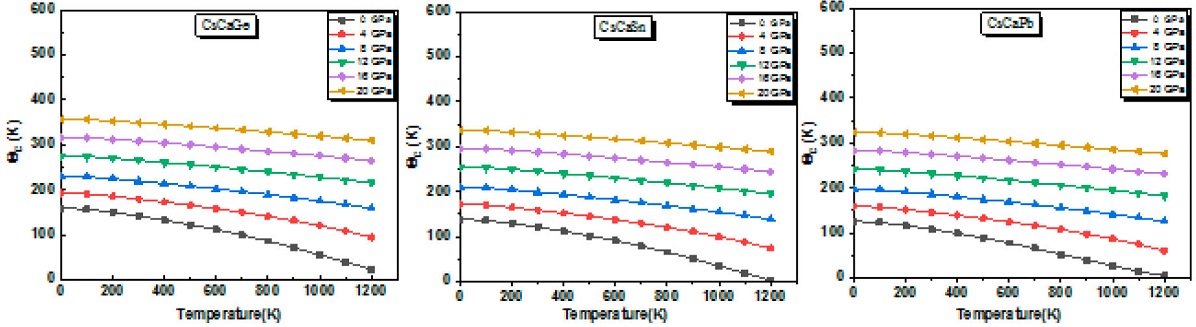
The Debye temperature (θD) is the most important parameter in the (QHDM), which is related to many important physical properties, such as melting temperature, elasticity, specific heat and lattice vibration [62]. The curves of θD against temperature at various pressures are shown in Fig. 9. At a given pressure, as the temperature increases, the θD decreases close to linearly. However, at a given temperature, θD increases for increasing pressure. Furthermore, one can see that the Debye temperature plots show quite similar features as compared with that of bulk modulus as both are indicators of material hardness.
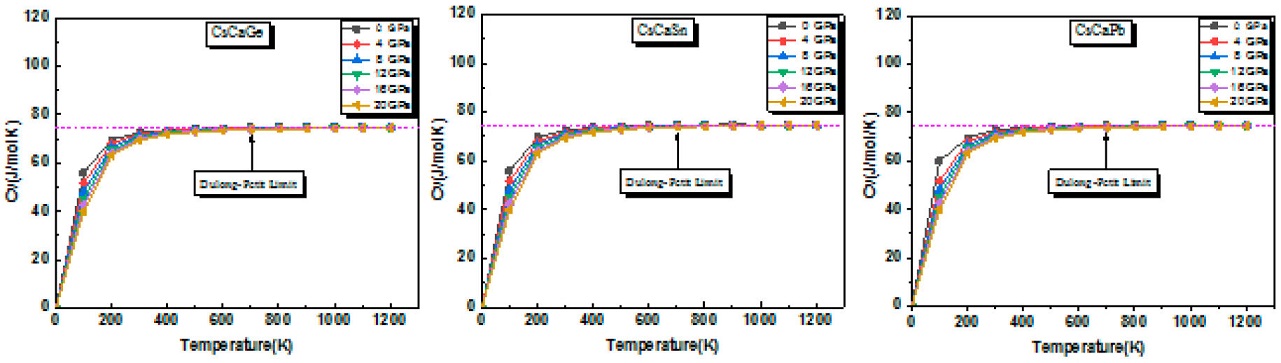
The heat capacity (Cv) of any compound is a significant factor that provides information about lattice vibration and phase transition. Figure 10 shows the effect oftemperature on Cv at various pressures. The result confirms that the increase of pressure has no influence on the heat capacity values. It is shown that Cv increases steeply with the initial 0-300 K temperature range since Cv obeys T3 law at low temperatures [63]. At 300 K and 0 GPa conditions, Cv values approache approximately 71.95, 72,60 and 73 J/mol-1K-1 for CsCaGe, CsCaSn and CsCaPb, respectively. From 300 to 700 K, Cv increases slowly with temperature. At temperature limit (T = 800 K), no more rate increase is observed and its value approaches approximately 75 J/mol-1K-1 for CsCaZ (Z= Ge, Sn and Pb) compound. As a result, the heat capacity is quite close to the Dulonge Petit classical limit, common phenomena in all solids [64].
Additionally, we studied the variation of the thermal expansion coefficient (α) as a function of temperature at different constant pressures. It can be noted from Fig. 11 that it increases sharply in the temperature range 0-300 K, followed by a gradual increase and beyond 300 K, particularly in high-pressure regions; it increases almost linearly with a low slope. As an instance, at room temperature, the coefficient (α) decreases only a (from 22.14 to 5.62 x 10-5/K for CsCaGe compound), whereas, it increases from 0 to 78.5 x 10-5/K) when the temperature is raised from 0 to 1200 K fixing pressure to 0 GPa.
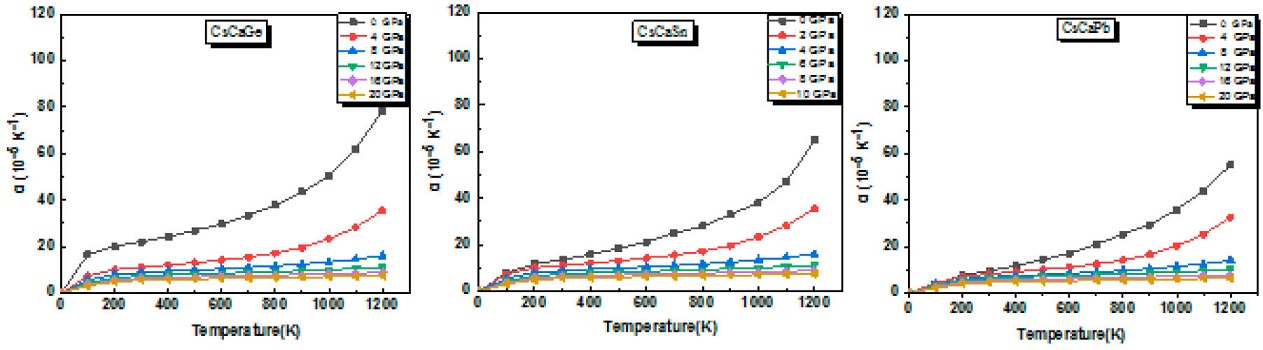
Figure 11 Thermal expansion coefficient (α) as a function of temperature for different pressure values.
Knowledge of Entropy (S) provides essential insight into the vibrational properties of a material leading to the understanding of the many performance applications of devices such as heat engines, refrigerators, and heat pumps. Figure 12 shows entropy as a function of temperature for different pressure values.
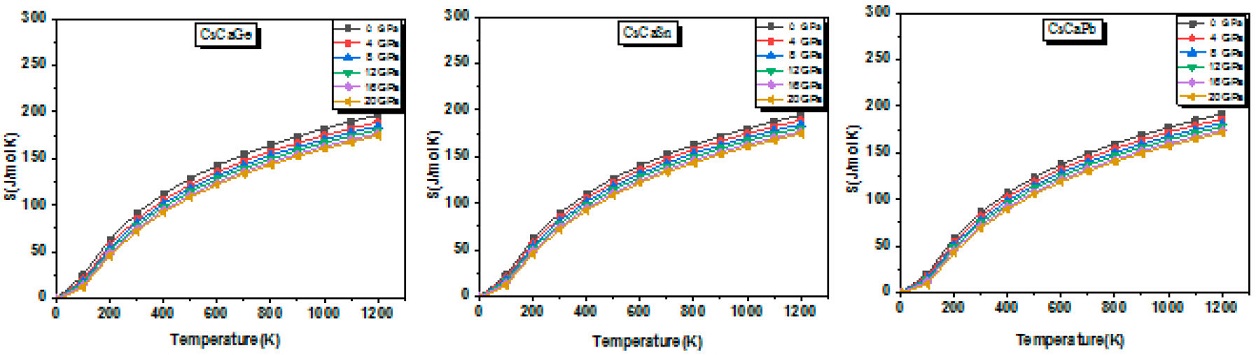
Figure 12 Thermal expansion coefficient (α) as a function of temperature for different pressure values.
It can be noticed from the plot that the stable equilibrium state corresponds to the starting curves values (S = 0) at 0 K and 0 GPa. In addition, as temperature increases, entropy increases exponentially and at the same time increases with pressure above 0 K. Furthermore,the calculated entropy values of 91.44, 89.34 and 86.33 J/mol-1K-1 for CsCaGe, CsCaSn and CsCaPb, respectively, indicate that CsCaGe is less ordered than CsCaPb and CsCaSn at 300 K and 0 GPa.
Finally, as temperature exceeds 800 K, entropy attains very high values. This is mainly due to the atomic vibrations for all compounds.
4. Conclusion
In this work, we examine the structural, elastic, electronic, magnetic and thermodynamic properties of the CsCaZ (Z = Ge, Sn and Pb) compound using the generalized gradient approximation (GGA-PBE). According to the calculated results, all three CsCaZ alloys crystallize into the α - phase ferromagnetic configuration in their respective ground states. The electronic band structure diagrams and density of states plots confirm the half-metallic nature with indirect band gaps of all materials and indicate that p-orbitals of the main group element (Ge/Sn /Pb) are the major contributors to the density of states. Morever, the total magnetic moment of 1 μB, which obeys the Slater-Pauling rule, is well maintained over a wide range of lattice parameter values 7.6-8.8 Å for CsCaGe and 7.8-8.8 A for CsCaSn/CsCaPb. In addition, simulations suggest the mechanical stability, elastic ductility and anisotropy of all compounds. The quasi-harmonic Debye model (QHDM) is also used to confirm the thermodynamic stability of the studied compounds. The bulk modulus, Debye temperature,heat capacity, thermal expansion and entropy under 0- 20 GPa pressure rangeand 0-1200 K temperature range are successfully calculated, lending credence to the current results regarding this new class of thermodynamically stable HHd0 alloys. Since there are no theoretical or experimental data for these compounds, we hope that the results reported in this work can serve as a reference for future experimental work. Finally, as CsCaZ (Z= Ge, Sn and Pb) compounds show suitable properties, it is expected that these materials will be good candidates for potential spintronic applications.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.

