











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkPACS: 34.20.Gj
1. Introduction
The study of the interaction energy among systems of six-carbon molecules and one zinc-dichloride molecule provides a potential energy curve indicative of the nature of the adsorbent material. Potential energy curves in two dimensions arise from the projection of potential energy surfaces in one plane. We consider that after the interaction, the carbon fragments obtained correspond to amorphous activated carbon (AC) either in planar or non-planar geometry. Planar corresponds to graphitizing AC, while adsorbent material represented by means of several surfaces fragments considered as hydrogen adsorbents.
Activated carbon use to be synthesized at different pore shape distributions by diverse methods at different temperatures and pressures1. Measurements of BET surface areas, SBET, give 1740 m2 g−1 for activated carbon powder (ACP) and 1970 m2 g−1 activated carbon fiber (ACF)2. The SBET of the ACF was 1.13 times larger than that of the ACP calculated.
In a previous research3 of burned coconut shells, the carbon obtained was activated using zinc dichloride [ZnCl2]. BET surface area measurements provided 208, 342, 452, 452 [m2/g] at 600, 700, 800, 900 [°C] respectively. The corresponding pore size diameters were 15, 14.8, 14.5, 14.5 [Å] obtained. Activated carbon with ZnCl2 as activating agent also was found to have pore size diameter: 17.6-18.4 Å4. According to the micropore size distributions (MPSD) measurements of activated carbón5, the most populated results are around 9 Å at the interval 6 to 18 angstroms, on which two different types of carbon materials were prepared: (i) chemically activated carbons prepared from an anthracite and a bituminous coal using KOH as activating agent, and (ii) physically activated carbon fibers prepared from petroleum pitch and coal tar pitch-based carbon fibers, by activation with CO2 and steam. Finally, the pore size distribution of activated carbons from adsorption data of different adsorbates was estimated by several methods6; particularly in the adsorption of nitrogen the range is 6-10 Å where their Eq. (9) lead them to the highest values of the average micropore width, while their Eqs. (13) and (14) lead them to the lowest ones.
Activated carbon is broadly defined to include a wide range of amorphous carbon based materials prepared in such a way that they exhibit high degree of porosity and an extended surface area7. Some researchers consider that the structure of activated carbon is quite similar to that of graphite composed of microcrystallites consisting of fused hexagonal rings of carbon atoms8. The spaces between the individual microcrystallites are called pores. Most of the adsorption takes place in micropores. Amorphous activated carbon is manufactured in many different forms: granular, fiber, and powder, as a few number of them.
The precise atomic structure of activated carbon is unknown9. The first carbon structures were found by means of pyrolysis, these carbons fall into two distinct classes: graphitizing and non-graphitizing. Choosing a particular activated carbon (carbonized from peat in an inert atmosphere at 500°C) and through HRTEM micrographs, growth of curved and faceted carbon sheets as blankets of (pentagonal, hexagonal and heptagonal) carbon rings networks displaying curvature can be observed, in some cases as bags enclosing much larger pores than in the fresh carbon10.
Diffusion of alkali ions on amorphous carbon by means of ab initio molecular dynamics for elucidating the interaction nature between K+/K and amorphous carbon was studied11 using seven different graphene sheets of hexagonal carbon rings, and considering them as models of amorphous carbon. A reaction mechanism of CF3I synthesis catalyzed by activated carbon was investigated with quantum chemistry methods using DFT12, obtaining: i) the adsorption configuration of some intermediates considering dehydrofluorination of CHF3 catalyzed by -COOH groups, which possess the highest barrier (ii) the difluorocarbene disproportionation over graphite (001) surface, and (iii) the corresponding pathway through TS1-2.
Random vs. realistic amorphous carbon models were confronted13 using measurements of high resolution microscopy and electron diffraction. The approach is to simulate a realistic carbon from an energetic model based on the tight-binding approximation. Then, a comparison of this carbon with the carbon obtained by randomly generated carbón atom positions, and concluding that the limit thickness for the weak phase approximation is 30% overestimated.
In the case of chemical activation several substances have been employed; however, the ZnCl2 has received special attention due to a high dehydrator effect that is conferred to raw material which attacks in solution to not extreme temperatures, promoting a fast and effective degradation of the cellulose material that makes up the majority of nuts and coconut shells14,15,16, coffee17, wood sawdust18,19, olive and prune seeds, walnut nut shells, paper, and organic materials20.
Active carbons are mainly and almost exclusively prepared by pyrolysis of carbonaceous raw material at temperatures lower than 1000°C. Activated carbon formed under the action of ZnCl2 is an adsorbent material proposed to study properties of hydrogen adsorption, and exhibits structural modification where the carbon is by means of ZnCl2 molecular arrangement activated. Structural properties might be for hydrogen adsorption-desorption processes considered.
Knowing that carbonization generates dehydrogenated carbon we consider hypothetical carbon molecules having zero content of hydrogen. And also knowing that carbon is the major constituent of active carbons7, and is present to the extent of 85% to 95%, in this work hypothetical molecules of either six-carbon chains or polyyne HC6H or hexagonal carbon rings are confronted to one ZnCl2 molecule. Some mechanisms of the reaction involved in the activation of carbon chains and diamond-like carbon by ZnCl2 are also considered. The calculations of energy by means of exchange and correlation functional using dnd basis sets of functions were accomplished.
2. Modeling and computational methodology
In order to figure out about suitable arrangements, we achieved DFT calculations of geometry optimization on molecular systems [C6] n ZnCl2 or [HC6H] n ZnCl2 arising from the reaction of either n-hypothetical six-carbon (chains or rings) or n-polyyne with a ZnCl2 molecule respectively. A geometry optimization gives the dissociation energy of the interacting molecules. In order to know the size of the potential well, single point calculations in order to obtain potential energy curves are achieved.
This arrangement is in symmetry group C1 investigated, through DFT electronic structure program21, using PW91 exchange-correlation functional in the generalized gradient approximation (GGA)22,23. The calculations are all-electron and spin-unrestricted (different orbital for different spin). Open-shell systems are accomplished with unrestricted wave functions. For these molecules, we use a dnd double numerical basis with polarization functions, i.e., functions whose angular momentum is higher than that of the highest occupied molecular orbital in the free atom. The size of the dnd basis is comparable to 6-31G** basis sets. Diamond-like carbons are investigated through DFT-m-GGA-M06-L functional, dnd basis, DIIS, and smearing of 0.9 Ha.
Connectivity24 calculations according to DMol3 on no bonding to s- and f-shell scheme, bond type, and converting representation to Kekulé, for bond length tolerances from 0.6 to 1.15 Å were accomplished in some cases of graphitic carbon.
Area calculations have been carried out by inserting triangles in each amorphous carbon ring and using the Heron formula:
3. Results
First of all, calculations in order to get credibility for using DFT in small systems are accomplished. Then, either polyyne or linear carbon, hypothetical six-carbon, and hexagonal carbon rings without hydrogen are confronted to zinc dichloride in order to obtain equilibrium energies. In spite of we are considering carbon molecules as hypothetical, we mention the kind of hybridization they correspond. All the results are in the following sections obtained.
3.1. Carbon-carbon interaction
In order to get a better idea of the order of magnitude of the approach of this methodology, carbon-carbon interaction has been calculated. A geometry optimization provides the minimum (1.3Å, -161.863 kcal/mol), which corresponds to the equilibrium point. The whole reaction pathway is shown in Fig. 1; where 156.3 kcal/mol as dissociation energy is obtained. While in the ground state
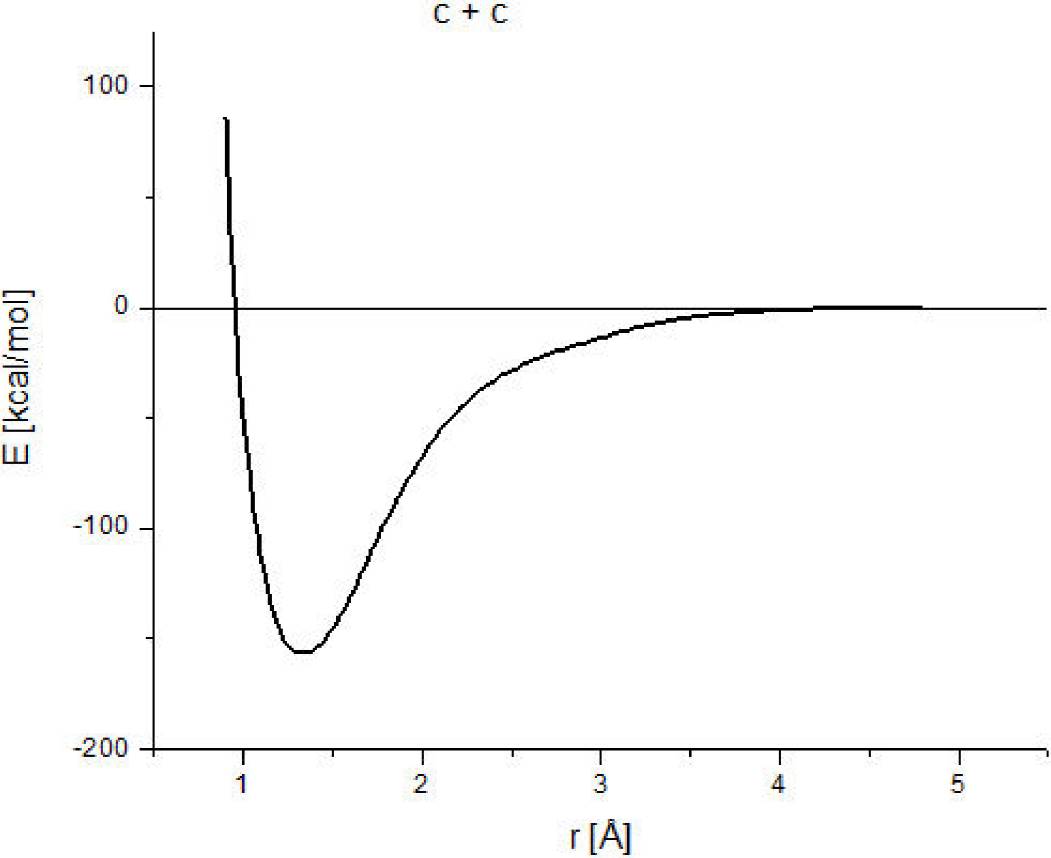
Figure 1. Here we observe the pathway of reaction or the potential energy curve of the carbon-carbon interaction. The bond strength is 156.3 kcal/mol.
Zinc and chlorine interaction energy with the same methodology was also calculated. The resulting dissociation energy of the ground state 50.53 kcal/mol agrees in 4.3% of difference with experimental25 value
In addition the geometry optimization of Cl - Cl interaction was carried out. These create the molecule Cl2 at 2.023 Å of separation and the resulting dissociation energy of the ground state 56.802 kcal/mol agrees with the experimental value
3.2 Hypothetical (-C≡C-)3 plus ZnCl2 interaction
Sequences of input-output molecular arrangements are shown in Fig. 2. Table I exhibits the equilibrium energies of the corresponding geometry optimization of this system and the bond strength. The first three parts (a, b and c) of Fig. 2 correspond to one hypothetical six-carbon chain alternating single and triple bonds (-C≡C-)3 and interacting with one zinc salt molecule in sp molecular hybridization. The only one difference in the input in each case is the initial separation among the reactants: a) 0.702 Å, b) 1.221 Å and c) 2.000 Å. The output exhibit strong differences: a) one bond of zinc salt decreased from 2.127 Å to 2.106 Å and the other increased until 5.312 Å; the reactants started linear and ended bent by 143° of the carbon chain molecule, and 129.048° of the zinc salt molecule. b) The zinc salt ended occupying the hydrogen places forming a whole linear chain molecule. c) The separation among the reactants increased from 2 Å to 3.85 Å indicating repulsion, and the linearity 180∘ changed to 177.4° for carbon chain and 174.105° for zinc salt.

Figure 2. A sequence of input - output molecular arrangements are shown. a) b) and c) correspond to one six-carbon chain interacting with one zinc salt molecule. And d) e) and f) correspond to two six-carbon chain interacting with one zinc salt molecule.
Table I. Equilibrium distance and energy along with the energy at infinity allow the calculation of the bond strength as

The last three parts (d, e, and f) in Fig. 2 correspond to two hypothetical six-carbon chains 2(-C≡C-)3 interacting with one zinc salt molecule where the distance among the reactants is shown in the input of Fig. 2. The output exhibits strong differences: d) the ClZnCl angle changed from 180° to 172.393°, the repulsion is from 1.1 Å until 4.166 Å, and carbon planar molecule tend to form six-ring. e) the ClZnCl angle changed from 180° to 138.530°, the repulsion allows the whole system 2(-C≡C-)3ZnCl2 as a planar molecule. f) the ClZnCl angle changed from 180° to 175.848°, the repulsion is from 0.853 Å to 4.057 Å, and carbon planar molecule tend to form one six-ring and one eight-ring.
3.3. 2C6 hypothetical chains plus Zn interaction
The molecular hybridization is mixed sp 2 and sp 3 for carbons in the body and at the end of the chain, respectively. Considering the molecular arrangement of one metal atom (Zn) between two parallel linear hypothetical six-carbon chains 1.54 Å separated as shown in Fig. 3a, a binding energy -1688.930 kcal/mol outcome from this geometry optimization of 64 steps with a Zn atom near to two crossed carbon chains as in Fig. 3b, where the whole system corresponds to a non-planar geometry arrangement. This result indicates that carbons arrangement corresponds to amorphous carbon.

Figure 3. a) Input: Zinc atom between two linear hypothetical six-carbon chains. b) Output: a non-planar C6ZnC6 optimized structure. It is clear an amorphous tendency of carbons arrangement in this geometry optimization. c) Potential energy curve calculation of single point where the minimum corresponds to the dissociation energy of (2C6)Zn molecule.
A potential energy curve between two six-carbon-chains and one Zn atom was determined. Calculations at 1-7 Å distances between one carbon atom in the molecule and one zinc atom as observed in Fig. 3c were achieved, by using small distance increments step by step with the purpose of determining the possible existence of adsorption. Adsorption due to the depth 68 kcal/mol of the potential well was found.
The interaction of two six-carbon chains with zinc atom is studied. First, the interaction energy values present a repulsive effect among these molecules at different separation distances: 2-3.1 Å. Second, they present an attractive effect from 3.1 to ∼7 Å of separation distances indicating that there is an adsorption of the Zn atom on two six-carbon chains. Then, the 2C6Zn molecule is formed at the minimum (-68 kcal/mol, 3.1 Å) of the pathway (see Fig. 3c, Table II).

In addition since zinc is an acid (acceptor)26 receiving an electron pair in its lowest unoccupied molecular orbital (LUMO) from the highest occupied molecular orbital (HOMO) of a base (donor). That is, the HOMO from the base and the LUMO from the acid combine with a bonding molecular orbital, which in our case corresponds with the orbital 102-HOMO and 103-LUMO, for Fermi Energy -5.107 eV, in the A irreducible representation of the C1 symmetry. The total number of valence orbitals is 192. The orbital occupation is 49 A (1) alpha and 49 A (1) beta, and 2.00 alpha electrons in 4 orbitals plus 2.00 beta electrons in 4 orbitals, for a total 102.0 number of electrons; and a binding energy -1688.930 kcal/mol.
3.4. 2C6 hypothetical chains plus ZnCl2 interaction
The hypothetical carbon chains have mixed sp 2 and sp 3 molecular hybridization. The molecular arrangement of one ClZnCl linear molecule between two parallel linear hypothetical six-carbon chains 1.54 Å separated as shown in Fig. 4a is for starting a geometry optimization considered. A binding energy -1891.068 kcal/mol result from this geometry optimization of 105 steps with a ZnCl2 non-linear molecule near to two crossed carbon chains, where the whole system corresponds to a non-planar geometry arrangement as seen in Fig. 4b. Another orientation of this Figure exhibits 1.36 Å instead of the initial 1.54 Å in one extremity, a bonding crossing in the other with a new bond 2.508 Å, and 1.735 [Å] of molecular separation (Fig. 4c). With this result it is clear that carbons arrangement is neither ring nor linear chain, certainly with porosity.
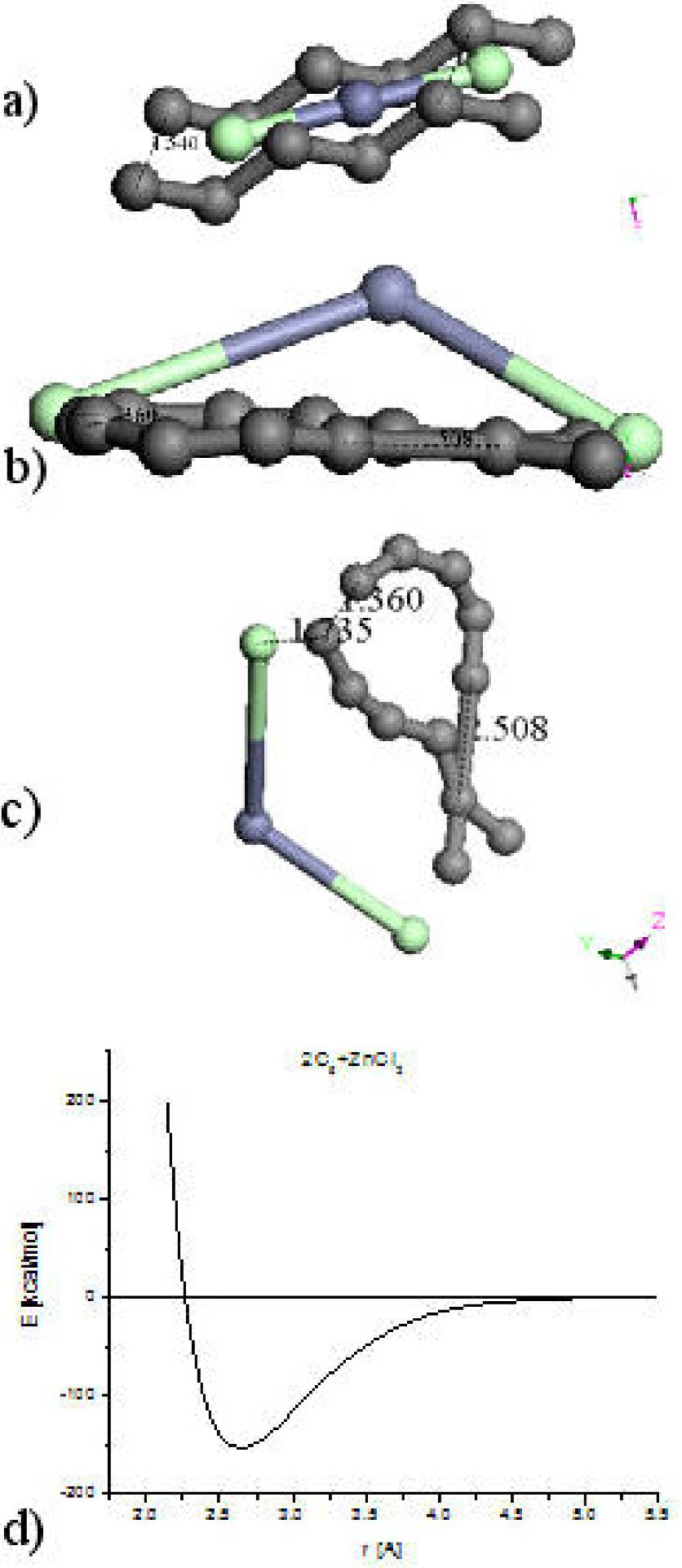
Figure 4. a) Input: Zinc atom into two chlorine atoms plus two 6-carbon linear chains. b) Output 1: a non-planar (2C6)ZnCl2 optimized structure, where 2C6 molecule is planar. c) Output 2: It is clear an amorphous tendency of carbons arrangement in these two geometry optimizations. d) The optimization energy calculation means an adsorption of amorphous activated carbon on Zinc salt at the minimum of the potential energy curve calculation of single point.
Initially, the interaction energy values present a repulsive effect among these molecules at different separation distances: 2-2.6 Å as in Fig. 4d. Next, the two six-carbon chains system and the molecule containing hydrogen present an attractive effect from 2.6 to ∼4.5 Å of separation distances indicating that there is an adsorption of the ZnCl2 molecule on the two six-carbon chain. Then, the [2C6]ZnCl2 molecule is formed at the minimum (-153.5 kcal/mol, 2.64 Å) of the pathway (see Fig. 4d).
The linear ZnCl2 molecule between two 6-linear chains (see Fig. 4a) was also optimized, obtaining [2C6]ZnCl2 as observed in Figs. 4b and 4c in different orientations.
The potential energy curve among two six-carbon-chains and a ZnCl2 molecule as in Fig. 4d was determined. Calculations at 1-10 Å distances between these two molecules were achieved, as observed in Fig. 4c, by using small distance increments with the purpose of determining the possible existence of an adsorption. An adsorption due to the depth 150 kcal/mol of the potential well was found.
The HOMO from the base and the LUMO from the acid combine with a bonding molecular orbital, which in our case corresponds to the orbital 136-HOMO and 137-LUMO, for a Fermi Energy of -5.222 eV, in the A irreducible representations of the C1 symmetry. The total number of valence orbitals is 228. The orbital occupation is 66 A (1) alpha and 66 A (1) beta, and 3.00 alpha electrons in 4 orbitals plus 3.00 beta electrons in 4 orbitals, for a total 138.0 number of electrons; and a binding energy of -1891.068 kcal/mol.
A small change on the position of the zinc salt linear molecule among the two carbon chains (Fig. 5a) was applied, and another geometry optimization shown in Fig. 5b was found, consequently the morphology of the activated carbon is different to the previous one; however, it is still amorphous. The relevant fact is that the potential well is 175 kcal/mol of depth (see Fig. 5c, Table II), which is greater than the previous one.
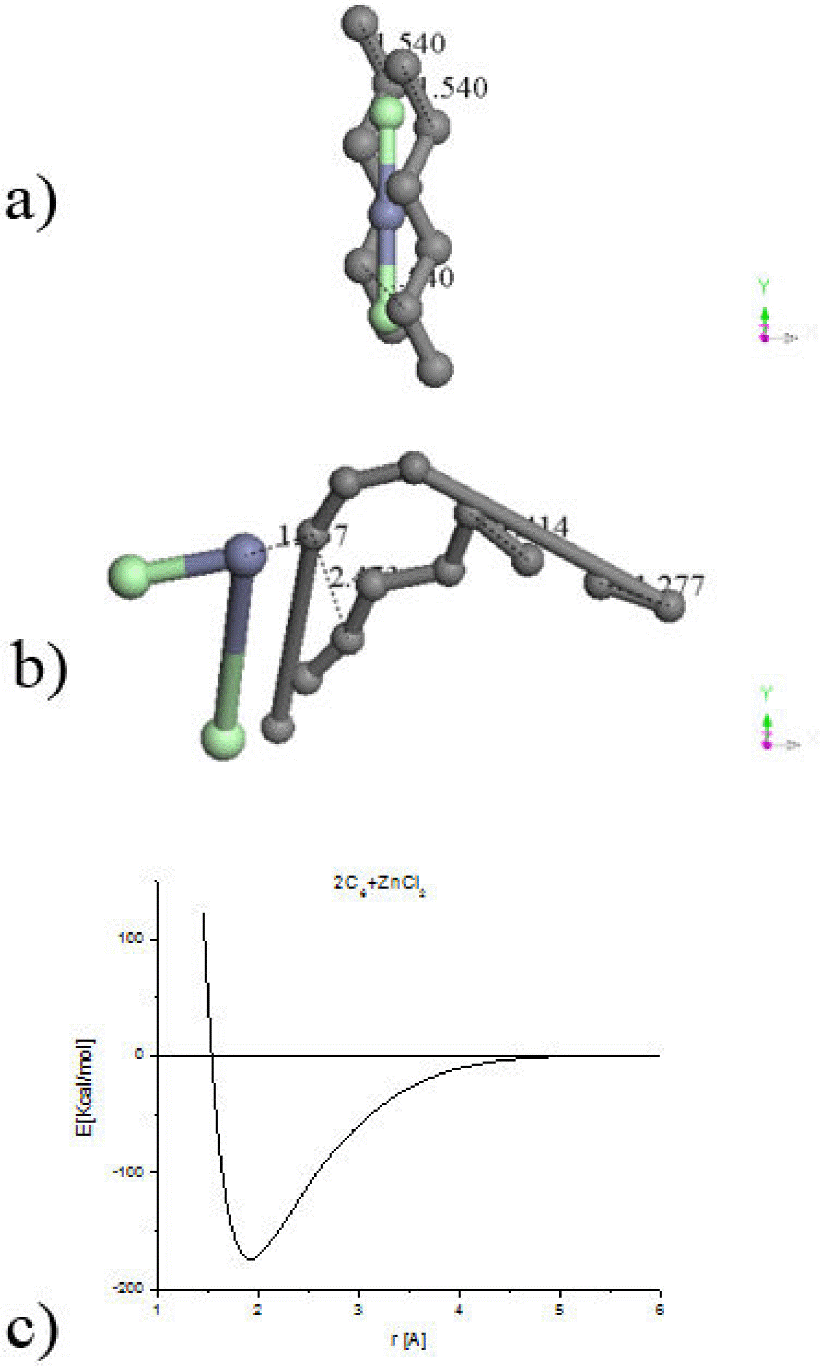
Figure 5. a) Input: Zinc atom into two chlorine atoms plus two 6-carbon linear chains at a small change of Fig. 4a, b) Output 1: a planar (2C6)ZnCl2 optimized structure. c)We observe the potential energy curve with 175 kcal/mol of well depth.
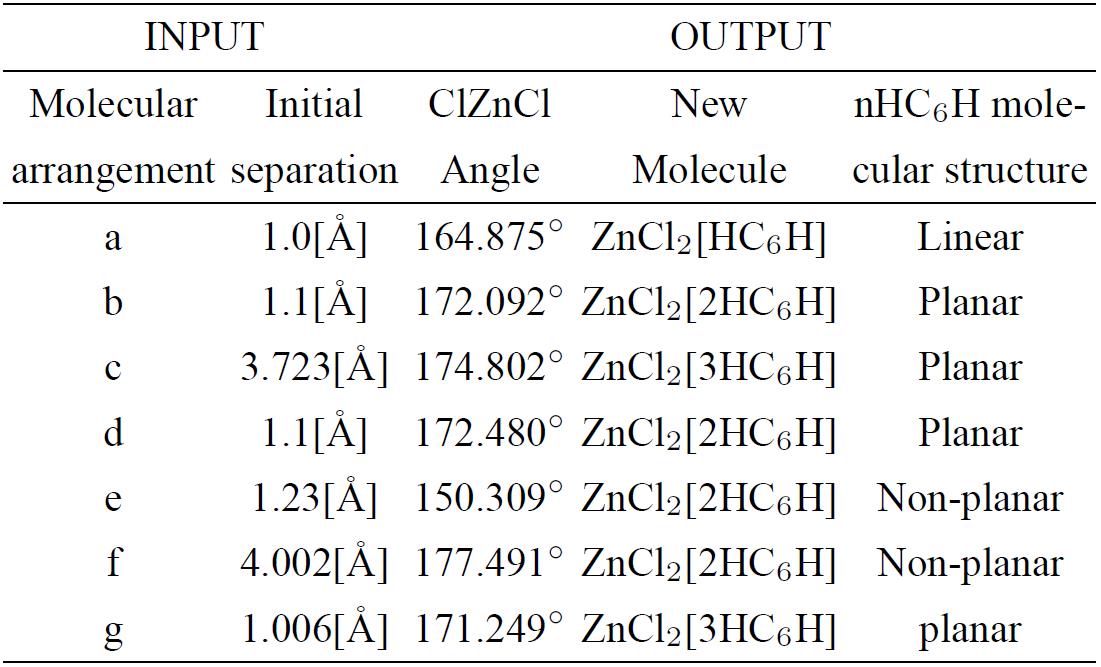
Table III. Initial separation is in angstroms and ClZnCl angle is in degrees of the input-output optimization geometry shown in Fig. 6.
3.5. Polyyne carbon chains plus ZnCl2 interaction
Previously, hypothetical single bond carbon chain dehydrated and dehydrogenated has been chosen in order to see its morphology. In this case the bond strength of carbon-chains with zinc (Fig. 3c) is around half of that for carbon-chains with zinc-salt (Fig. 5c). In this address, an example of a known hydrogenated linear carbon chain is polyyne carbon, which is an allotrope of carbon that has a chemical structure H(-C≡C-) n H as a repeating chain, with alternating single and triple bonds27 and hydrogen at every extremity as any member of the polyyne28 family HC 2n H corresponding to sp hybridization atoms.
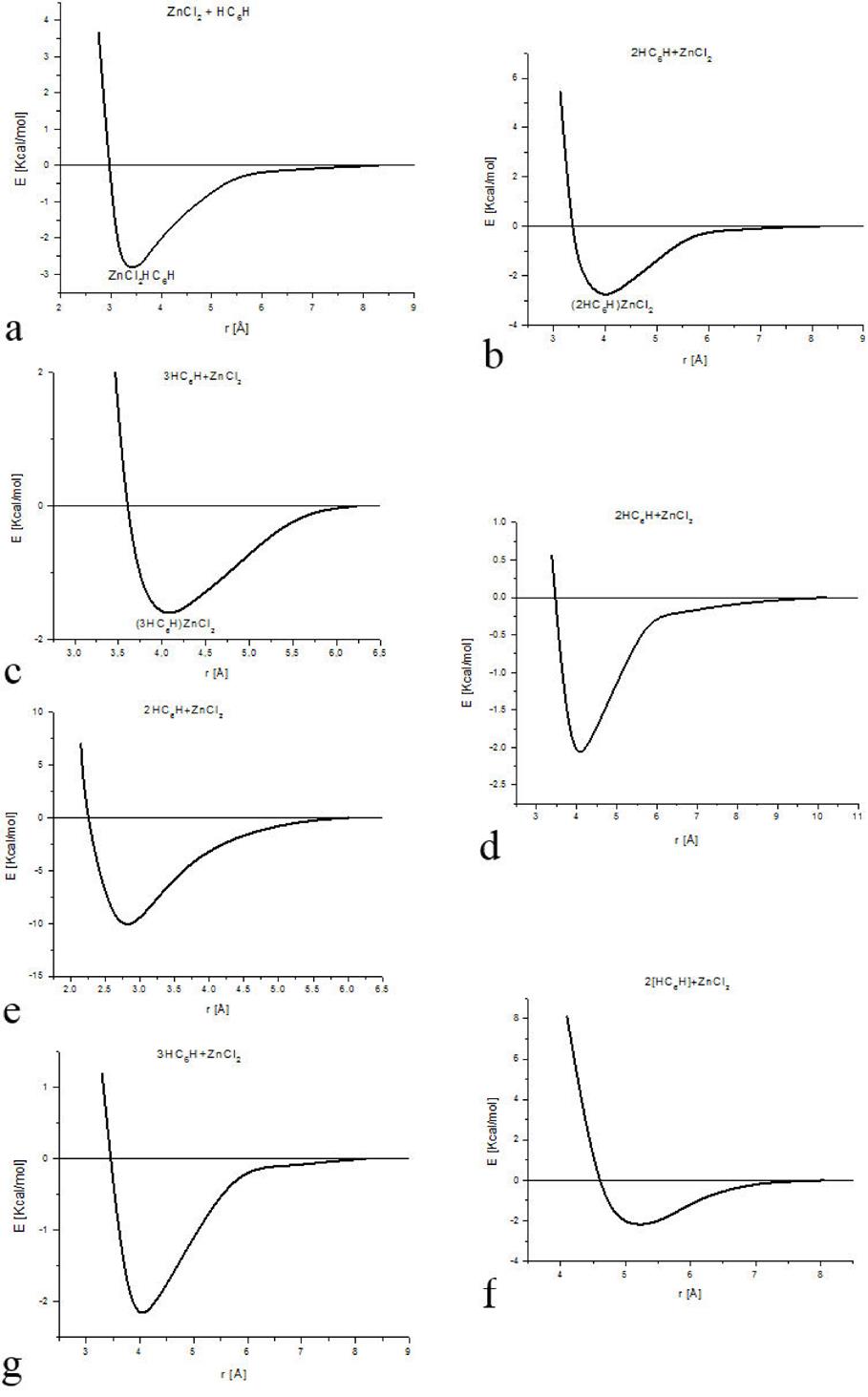
A geometry optimization is applied on different arrangements of the zinc salt molecule interacting with H(-C≡C-)3H polyyne molecules (See Fig. 6 and Table III). The output of Fig. 6 shows the geometry of the activated carbon, which we are to describe to continuation: a) A linear carbon chain, b) Flat carbon chain molecules separated 4.033 Å, c) Three curvilinear carbon chain separated at the extremities and forming a weird six carbon ring in the center, where the whole carbon system is located in one geometric plane, d) Two carbon chain molecules separated in one side and forming an eight carbon ring two bonds open in the other side, e) non-planar carbon chain molecules in an S geometric form, f) two parallel semi-cylindrical carbon chain molecules g) two carbon molecules with zinc salt form four six-carbon rings. The graphs of the potential energy curves of Fig. 6 are exhibited in Fig. 7, and the corresponding equilibrium energies are given in Table IV.
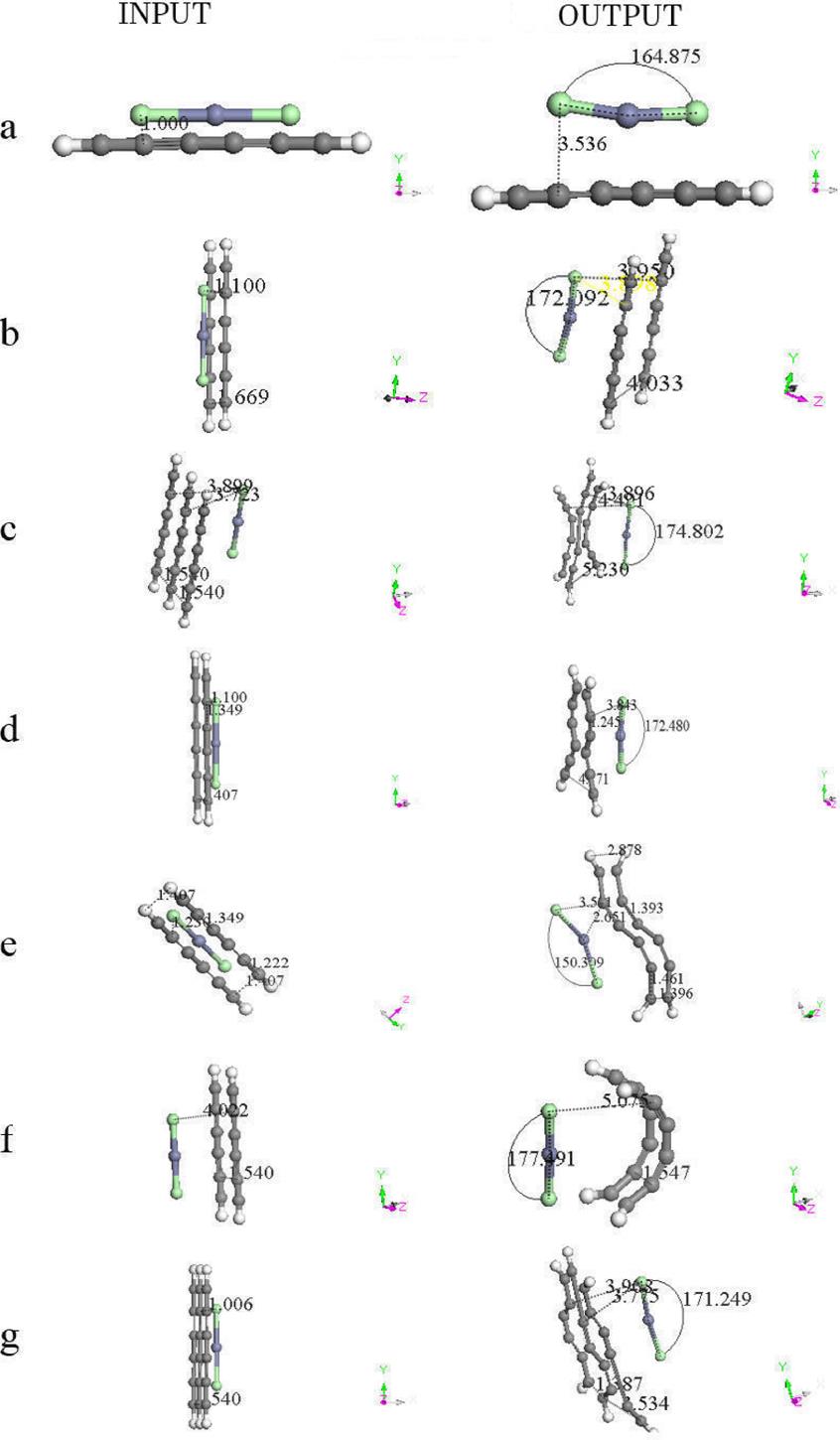
Figure 6. Input - Output scheme of the optimization energy corresponding to zinc salt which is near to one, two or three six carbon chains. The latter is forming new non-planar molecular structure in most of the cases. See Table III.
Table IV. Equilibrium distance Re and equilibrium energy Ee corresponding to the minimum of each potential energy curve in Fig. 7. Energy in [Kcal/mol], and distance in [Å].

3.6. Hexagonal carbon rings plus ZnCl2 interaction
We start calculating DFT geometry optimization of one hexagonal carbon ring with only one zinc metal atom in its center. The hybridization of ring carbons is sp 2 . After the optimization, a 35.10% area growth or AO= 1.3510AI for AI= 6.1616A2 (see Figs. 8a) was calculated, where AI is the input area and AO is the output area; while using one linear zinc salt (ZnCl2) molecule perpendicularly to the plane of the ring produces 43.53% area growth or AO= 1.4353AI as observed in Figs. 8b. The largest part of the growth comes from the metal atom, however zinc salt gives greater growth than zinc and can be removed from carbon by washing with water, while zinc stays adsorbed in carbon molecules. Table V provides binding energies, and pore size diameters in a whole output system PSDW and in the biggest ring PSDR for every output illustration in Fig. 8.

Figure 8. Input - Output scheme of geometry optimization corresponding to zinc salt acting on a specific hexagonal carbon ring as observed. See also Table V.
Table V. Binding energy and pore size diameter (PSD) where PSDW means whole system and PSD R means the biggest ring in Fig. 8. Energy in [Kcal/mol], and pore size diameter in [Å].

*This case corresponds to psdsl
In the rest of this work we will use sheets of hexagonal carbon rings at interaction with linear zinc salt as input to the simulation, where zinc is located in the middle of the linear optimized zinc salt, in order to calculate geometry optimization of the whole system.
Adding one hexagonal carbon ring to the previous one as input (see Fig. 8c) for calculating the geometry optimization take us to the output in this case. It is very clear the amorphous form of the activated carbon in this result, in which the zinc salt passed through one bond of a carbon ring until a distance of 1.318 Å, and the equilibrium energy -1609.841 kcal/mol corresponds to chemisorption29. The initial form of these two rings was seriously distorted at the end of the optimization. It can be observed that carbons have a tendency to form a circle due to the position in which the zinc salt is located at the beginning. Actually the area grew 31.2% because AO= 1.312AI for AI= 12.3232A2, which is lower than the previous one. Connectivity calculation24 exhibits clearly the insertion of zinc dichloride into a new carbon ring.
A new input is to add another hexagonal carbon ring as in Fig. 8d, then after the geometry optimization a polygonal carbon ring of 14 sides is observed. This confirms a tendency of carbon atoms to form circle rings. Furthermore, the area calculated using Heron formula is 25.56 Å2. Using this as a circle area (πr 2 ), the pore size diameter can be approximated as D= 2(A/π)1/2= 5.8 Å. Actually the diameter measured is 5.77 Å. In this case AO= 1.3827A I , where AI= 18.4848 Å2. Connectivity keep invariant these calculations, and the new molecule corresponds to one carbyne case30.
A symmetrical addition of another hexagonal carbon ring in the input as in Fig. 8e provides a polygon of 18 sides after the geometry optimization in the output of this Figure. This is the best circle carbon ring obtained here with three bonds inside of the same, as disk morphology. The triangle area is AT= 42.18389 Å2, while the circle area is AC= 43.00840 Å2. They are almost the same. Here AO= 1.71156AI which means ∼71.15% of growth. In this case the growth of the ring with zinc salt inside is AO= 4.4281AI for AO= 6.1616 Å2 with a pore size of 5.894 Å, while in the previous case it is AO= 3.0799AI. Connectivity24 calculation provides an 18-carbon ring sharing single and triple bonds, which corresponds to one carbyne molecule30.
Completing 5 hexagonal carbon rings as in the input of Fig. 8f, and after the geometry optimization two decagonal carbon rings were obtained in the output, with one bond in the middle of each one, and connected by two bonds, where AO= 1.3524AI for AI= 30.81 Å2. It is clear in this case that linear zinc dichloride molecule remains as a salt molecule because it is not broken or separated. The potential energy curve has a minimum in (0 [Å], 16 Kcal/mol), obtained in the x direction and taking vertically this molecule. Then adsorption is located in the threshold among physisorption and chemisorption for a potential well depth around 16 Kcal/mol. Connectivity calculation exhibit two 10-carbon ring alternating single and triple bonds, separated equidistantly from linear zinc dichloride. Another form of amorphous activated carbon comes out when six hexagonal carbon rings were joined, as in the input shown in Fig. 8g. In this case the area calculated in the output is lightly compacted: AO= 0.999256348AI for AI= 36.969585 Å2. Connectivity calculation leads us to observe zinc atom bonded to three carbon atoms, and two carbon atoms stayed inserted in the zinc dichloride bonds leaving dichloride molecule in the carbon atom at the end. This new carbon molecule contaminated by zinc and chlorine has single, double, and triple bonds.
The last case here exhibited is when 7 hexagonal carbon rings were joined as in the input shown in Fig. 8h. After the geometry optimization the area in the output suffered a greater compression than in the previous case: AO= 0.909908362AI for AI= 43.1312 Å2. The growing and compressing of the area after the geometry optimization is a clear indication of the existence of very different kind of pore size distribution in the amorphous activated carbon.
In case of stacking sheets of carbon rings, the initial point corresponds to 12 carbon atoms of two hexagonal carbón rings in face to face orientation, where zinc salt is perpendicularly located among both rings as observed in Fig. 8i, and from the geometry optimization shown in the output A = 32.448 Å2 is calculated. Then 6.4276 Å2 is the pore size diameter approximately, which for a small system like this, with just one zinc salt molecule and 2 hexagonal carbon rings is a good result. After connectivity calculation chlorine atoms stayed bond to the lowest carbon atoms, while zinc is bonded to the highest carbon atoms in Fig 8i.
One stacking of two sheets case is exhibited in Fig. 8j. These are 26 carbon atoms showing the particular amorphous activated carbon form after geometry optimization of a dimmer of two sheets of three carbon rings in face to face orientation. The area of the biggest ring after optimization is ∼15 Å2 which corresponds to ∼4.37 Å of pore size diameter approximately. Then the area of this ring grew ∼2.43 times. Connectivity calculation lead to ∼5.4 Å of pore size diameter of the biggest ring, and to a carbon molecule contaminated by zinc, and also by chlorine in different places, because zinc share three carbon atoms while chlorine share only one carbon atom as if this were a hydrogen atom.
Another stacking of two sheets is exhibited in Fig. 8k. These are 48 carbon atoms showing activated carbon after geometry optimization of a dimmer of two sheets of seven carbon rings in face to face orientation. The input volume among graphitic carbon sheets is 150.959 Å3, and after optimization is 275.91 Å3 which corresponds to 8.08 Å of pore size diameter. Then the volume grew 82.8 % or AO= 1.8277AI. Connectivity calculation lead to separation of zinc salt molecule as ZnCl+Cl, and while the chlorine atom stayed between the two graphitic carbon sheets, the ZnCl molecule keeps far away from the closest graphitic sheet.
These two cases have in common the metal separation bond of the chlorine atoms, showing that carbon is a natural adsorbent of metals, and sending the chlorine atoms until the end. These two geometry optimizations, first of a dimmer of just one hexagonal carbon ring in face to face orientation with one zinc salt molecule, and second of a dimmer of hexagonal carbons also in face to face orientation, with zinc salt crossing each sheet of three hexagonal carbon rings, were able to separate the bonds of zinc dichloride in a complex [2C13]ZnCl2 → ClC13ZnC13Cl and a complex [2C6]ZnCl2 → ClC6ZnC6Cl. While the first complex is non-planar, the second one is planar.
3.7. Diamond-like carbon plus ZnCl2 interaction
For activating diamond using zinc dichloride as activating agent, through a Geometry optimization, DMol3 m-GGA/M06-L functional is applied in order to build an optimized diamond-like structure, in the following steps. First the geometry of a C6 ring is optimized. Second, to the latter we added the missing four carbons for optimizing the whole sp 2 structure of a unit diamond. Third, the whole unit structure diamond of fourteen carbons is optimized. Finally an optimized zinc dichloride molecule is placed as observed in Fig. 10a,b as input-output of the geometry optimization searched. A potential energy curve when the activating agent cross this diamond-like molecule in x-direction as shown in Fig. 10c. A potential well depth around 20 Kcal/mol means chemisorption when zinc dichloride is inside the diamond-like molecule. When this activating agent is going out the maximum around 5 Å means a very strong chemisorption, and the minimum around 13 Å corresponds to a physisorption.
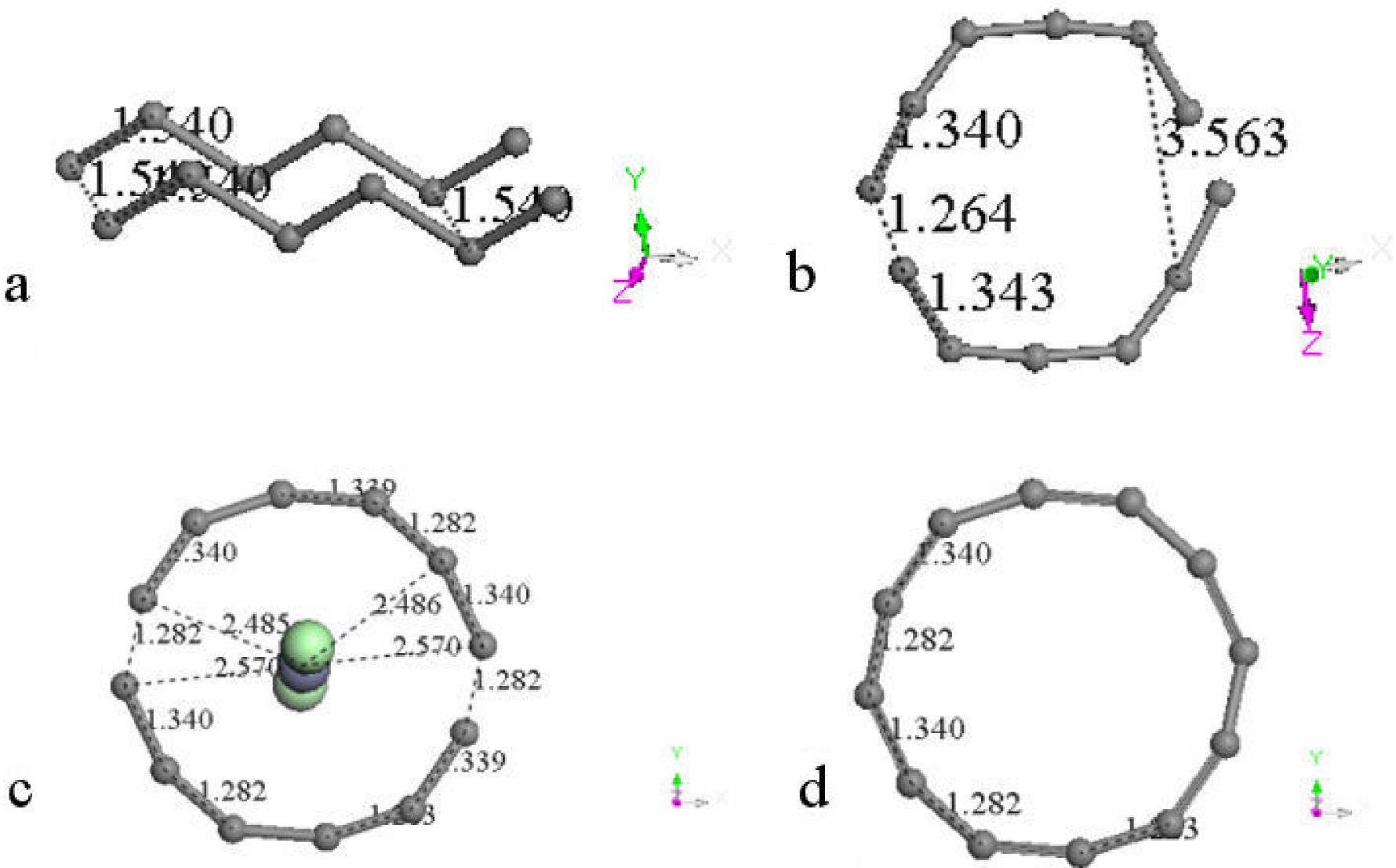
Figure 9. a) Two six carbon chains separated to a bond distance. b) These carbon chains change to a twelve carbon ring after geometry opyimization. c) Pore size area increase 4% after carbon activation with zinc salt. d) Connectivity of the activated carbon ring.

Figure 10. a) Input: Optimized diamond molecule. b) Output: Activated carbons of a diamond. c) Potential energy curve of the interaction Diamond + Zinc Dichloride.
Applying an average calculation to the input-output bond lengths of diamond-like molecule, ∼18% growing of activated carbon of diamond with zinc-dichloride is observed. Diamond formula is C14H24, with density 3.514 gr/cm331, and mass 192.34 gr, then diamond volume is 54.74 cm3, and activated diamond volume is 64.59 cm3. Graphite density is 2.266 gr/cm331, with sp 2 hybridization, then
ρ ad = 1.3141ρ g , and ρ ad = 0.847ρ d
where ρ ad is density of activated diamond, ρ g is graphite density and ρ d is diamond density. Therefore
ρ g < ρ ad < ρ d
in diamond there is sp 3 hybridization of four carbons, sp 2 of six carbons, and the last four carbon share all its free electrons. This means that 40% is sp 3 , and 60% is sp 2 .
4. Discussion
Our different geometry optimizations obtained for very similar cases take us to consider that PSD may appear in: i) inside carbon rings that we define as psdcr, ii) between graphitic sheet layers forming cavities that we define as psdsl, and iii) inside granular diamond-like carbon that we define as psdgd. In the psdcr case there are two results 3.8 Å and 7.4 Å of pore size diameter for those shown in Figs. 4c and 8e, respectively. In case of psdsl Fig. 8k provides the best 8.08 Å pore size diameter, very close to the most populated pore size distribution 9.0 Å diameter. In case of psdgd our system grew 18% only, with 4.98 Å of pore size diameter. Our best calculation of pore size diameter for one carbon ring is 8.02 Å, which is around the order of 55% of those experimentally measured for activating carbon using ZnCl2. We applied connectivity24 calculations in many cases of Fig. 8, due to unrealistic long bonds, and considering that connectivity and PSE methods produce similar results in chemisorption cases, and SCF calculations do not converge to build potential energy curves in most of these cases. Connectivity of ionic or covalent bonds allow to observe active carbon with zinc chemically adsorbed in Figs. 8c, 8h-8j. Then the orientation of the interaction can provide a big percentage of zinc inserted in activated carbon when using zinc dichloride as activating agent. Scattering data take us to average potential energy curves32,33 hiding information about physisorption or chemisorption, because the orientation of the interaction is not important when taking this kind of data.
The use of six-carbon chains in this model is due to graphite and graphene are hexagonal carbon sheets. One very good example is shown in Fig. 9, where two linear six-carbon chains after geometry optimization form one ring of twelve carbons with an Area of 18.39 Å2 and pore size diameter 4.84 Å (Fig. 9b). Activating this carbon ring with zinc dichloride provides a pore size diameter of 4.94 Å (Fig. 9c) with only 4.2% of growth, and close to the largest values obtained previously. We also see in Fig. 9d that calculating connectivity single and triple bonds appear, consequently this twelve carbon ring is strongly bonded with σ and π electronic configuration interactions.
Considering that ZnCl2 push carbon, we also propose that the equilibrium distance Re in Tables I and IV allows pore size diameters among 5.3 Å and 10.4 Å close to those measured previously. Our results have a close agreement with experimental micropore size distributions (MPSD) measurements of activated carbon around 9 Å and agree with the lowest values of average micropore width. We must note that Fig. 8e exhibits a disk form after geometry optimization. Due to this fact, it is easy to imagine more regular geometries as cylinders and spheres also formed when carbon is under the action of metal activating agents. In crude oil fields nickel is a natural activating agent of carbon molecules, and disks, cylinders or spheres have been widely detected34.
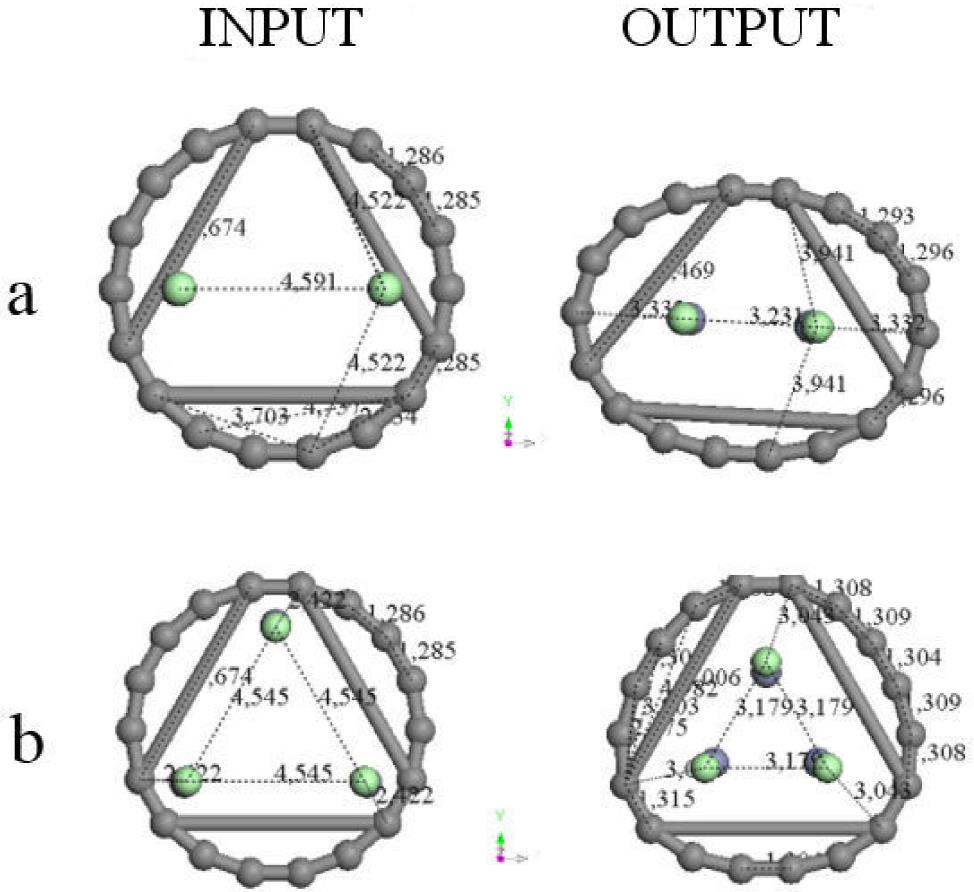
We applied two and three zinc salt molecules inside the 18-side carbon ring obtained after geometry optimization, and we applied geometry optimization again. Figure 11 exhibit these two cases. In the former case we only observe deformation as an ellipse of the 18-carbon molecule, because the area stays invariant. In the latter case we observe a small growing of the area, because the pore size diameter grew from 7.4 to 7.52 Å.
Zinc dichloride is a salt that can be washed from the activated carbon when the [2C6]ZnCl2 molecules are in contact with water, however zinc has been detected in activated carbon after its cleaning.
5. Conclusions
First, the tendency of linear chains after geometry optimization is to form aromatic rings; however, we used two linear carbon chains which after optimization changed to amorphous carbon. The latter occurs when they get in contact with zinc dichloride under the action of a geometry optimization. Second, there is a very strong potential well depth 175 kcal/mol among the twelve carbons and zinc dichloride, which is indicative of a chemisorption according to the range handled by Atkins29; however, in polyyne carbon case all the potential well depths are lower than 11 kcal/mol corresponding to physisorption. This means that even a small quantity of hydrogen, adsorption changes significantly. Activated carbon morphology depends on the carbon and on the methodology used.
Our best model provided a pore size diameter of 8.08 Å in agreement to experimental results for the average pore size distribution diameter. Improvements are still in development considering carbon chains (linear or rings) greater than six-carbon molecules, and more molecules of zinc salt or another activating agent. Linear Carbon interactions tend to form carbon rings after geometry optimization, and according to the case they stay either linear or planar or non-planar. We consider planar as graphitizing and non-planar as non-graphitizing AC.
A true formation of [2C6]ZnCl2 molecules are obtained at the potential well when one molecule of two six-carbon chains interacts with ZnCl2, which have to be washed in order to improve quality of activated carbon. Our results give evidence that at particular cases Fig. 8g, i, j, carbon stayed inserted in the bonds of ZnCl2 molecule, then zinc might appear in the composition of activated carbon using zinc dichloride as activating agent, despite washing.

