











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkThe term “triglyceride-rich lipoproteins” (TRLs) include a heterogeneous set of particles ranging from the recently secreted lipoproteins in the gut (chylomicrons) and the liver (very low-density lipoproteins [VLDL]), to their end products that result from their contact with lipolytic enzymes (mainly, lipoprotein lipase [LPL]). The chylomicron end products are known as remnants; the intermediate-density lipoproteins (IDL) are the corresponding ones for VLDLs. Some (remnants, IDLs, and the denser VLDLs) but not all TRLs are involved in the development of atherosclerotic cardiovascular disease1-3. As a result, clinicians should be able to discern in which cases TRLs should be considered as treatment targets (besides low-density lipoprotein [LDL]-cholesterol). The objective of this review is to discuss the mechanisms by which TRLs are involved in atherogenesis. Furthermore, we summarize the controversial aspects of the clinical approach and the treatment of cases with dyslipidemia explained by atherogenic TRLs.
POSSIBLE MECHANISMS OF ATHEROGENESIS INDUCED BY TRLS
TRLs, particularly the denser VLDL, IDL, and chylomicron remnants, can promote atherogenesis through several mechanisms (summarized in Fig. 1).
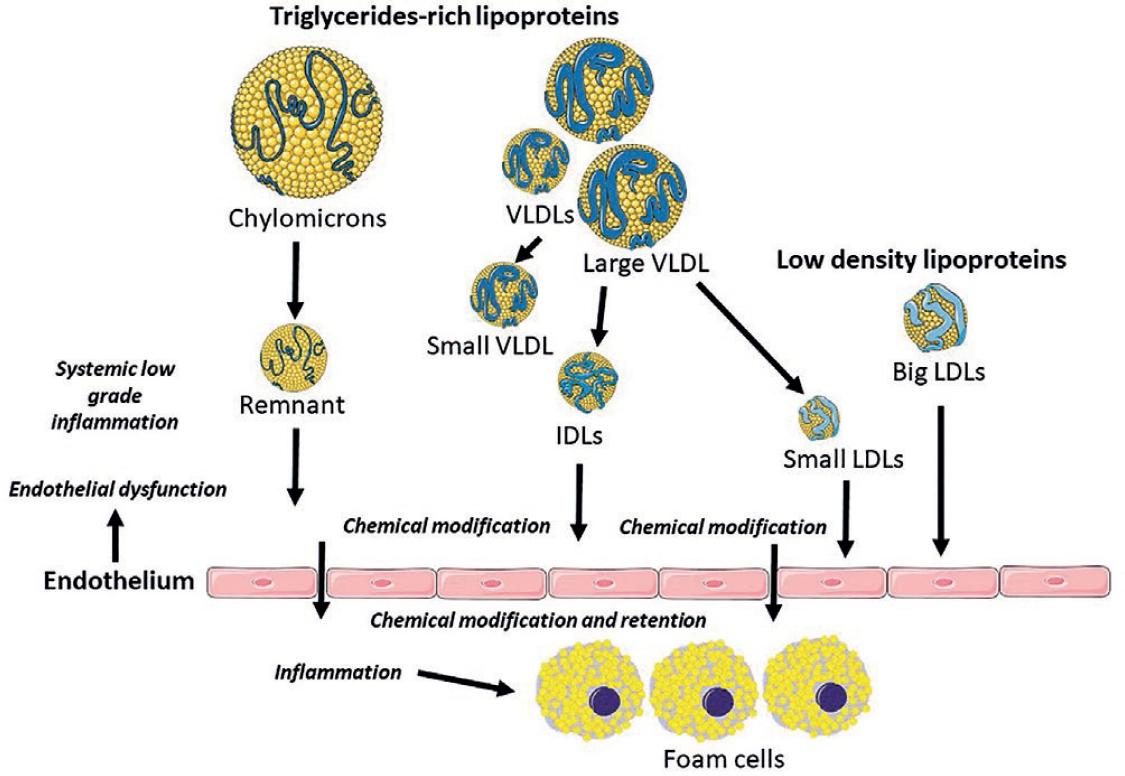
Figure 1 Triglycerides rich lipoproteins: direct and indirect mechanisms that explain their involvement in atherosclerosis.
VLDL accumulation promotes pro-atherogenic changes in other plasma lipoproteins (High-density lipoproteins [HDL] and LDL) by accelerating neutral lipid exchange reactions
VLDL particles are separated into two main classes: large, triglyceride-rich VLDL particles (50-80 nm in diameter and 70% triglyceride mass), called VLDL1, and smaller, denser particles (30-50 nm in diameter and 30% mass of triglycerides), called VLDL24,5. Diseases in which the hepatic secretion of VLDL1 is increased (i.e., type 2 diabetes mellitus and Familial Combined Hyperlipidemia) are characterized by the presence of hypertriglyceridemia, lower HDL-cholesterol concentrations and the formation of small and dense LDL4-7. In the context of high levels of the recently secreted VLDL1 particles, cholesterol ester transfer protein (CETP) activity increases, which mediates the transfer of cholesterol esters from HDL2 to VLDL1 and the reciprocal transfer of triglycerides from VLDL1 toward HDL28,9. This lipid exchange produces HDL2 enriched with triglycerides and VLDL enriched with cholesterol esters. Triglycerides-enriched HDL2s become a substrate of hepatic lipase, an enzyme that produces lipolysis of triglycerides and phospholipids leading to their conversion on smaller and denser HDL particles (known as HDL3 or “HDL remnants”) and lipid-poor apolipoprotein A-I (apoA-I), which is rapidly cleared from the circulation by the kidney10,11. The formation of small, dense LDLs is closely associated to this process. CETP facilitates the transfer of triglycerides from TRLs to LDL, resulting in increased affinity for hepatic lipase and hydrolysis of triglycerides. The resulting LDLs are denser, cholesterol-depleted, smaller in diameter with an increased affinity for the subendothelial space and a longer plasma half-life. The small dense LDLs (sdLDLs) predominate among LDL particles when plasma triglycerides are above 133 mg/dL12. In summary, the plasma accumulation of triglycerides-enriched VLDL1 causes atherogenic changes in HDLs and LDLs.
Physiologically, HDLs exert pleiotropic effects that prevent atherosclerosis13,14. They promote the reverse transport of cholesterol from peripheral organs (i.e., lipid-laden macrophages) to the liver to be excreted through bile and feces. In addition, HDLs have anti-inflammatory effects on vascular endothelial cells by decreasing the expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin. These molecules mediate the adhesion of circulating monocytes to the endothelial surface, one of the first steps in the sequence of events that culminates in the formation of atherosclerotic plaque. Furthermore, HDLs prevent LDL oxidation, mediated by several HDL components, including apoA-I, paraoxonase I, lecithin cholesterol acyl transferase, acyl hydrolase of the platelet-activator factor (PAF-AH), apoJ, apoF, and the phospholipid transfer protein. Other mechanisms by which HDLs reduce atherogenesis are protection against endothelial cell apoptosis, stimulation of endothelial cell repair, improved endothelial function by producing upregulation of nitric oxide (NO) synthase (eNOS), which increases vascular NO, and anti-thrombotic effects by decreasing the expression of tissue factor in endothelial cells.
The sdLDLs are more atherogenic than large buoyant LDLs due to multiple mechanisms12,15-19, schematized in figure 2. SdLDLs have a prolonged residence time in plasma as a result of their reduced affinity for the LDL receptor (LDLR), caused by an alteration in the conformation of the ligand region of apoB. Therefore, the probability to move to the subendothelial space is higher. Due to their smaller size, these particles can penetrate the endothelial barrier easily (1.5-1.9-times more compared to other LDLs). In the endothelial barrier's surface, there is a decreased concentration of sialic acid; consequently, its affinity for the proteoglycans of the arterial intimal matrix is increased, facilitating their retention in the subendothelial space and in the plaques. Furthermore, sdLDLs have a greater susceptibility to suffer oxidative modifications (perhaps caused by a decrease in their vitamin E content) and glycation (possibly due to a higher proportion of lysine residues being exposed on the surface of the particles and/or to their longer residence time). This increased susceptibility to glycation of sdLDLs occurs even in the absence of hyperglycemia. These changes increase the particles' affinity for the scavenger receptors of macrophages residents in the arterial intima. Macrophages will be saturated with cholesterol esters, and become foam cells, which is the initial lesion of atherosclerosis. In the endothelial cells, sdLDLs stimulate the secretion of plasminogen activator inhibitor-1 (PAI-1) and thromboxane, leading to increased thrombogenicity. In the smooth muscle cells of the artery wall, sdLDLs increase intracellular calcium concentrations, augmenting the vasomotor properties of the coronary arteries. Finally, these particles are enriched in atherogenic apoproteins (i.e., apoC-III) and the lipoprotein-associated phospholipase A2.
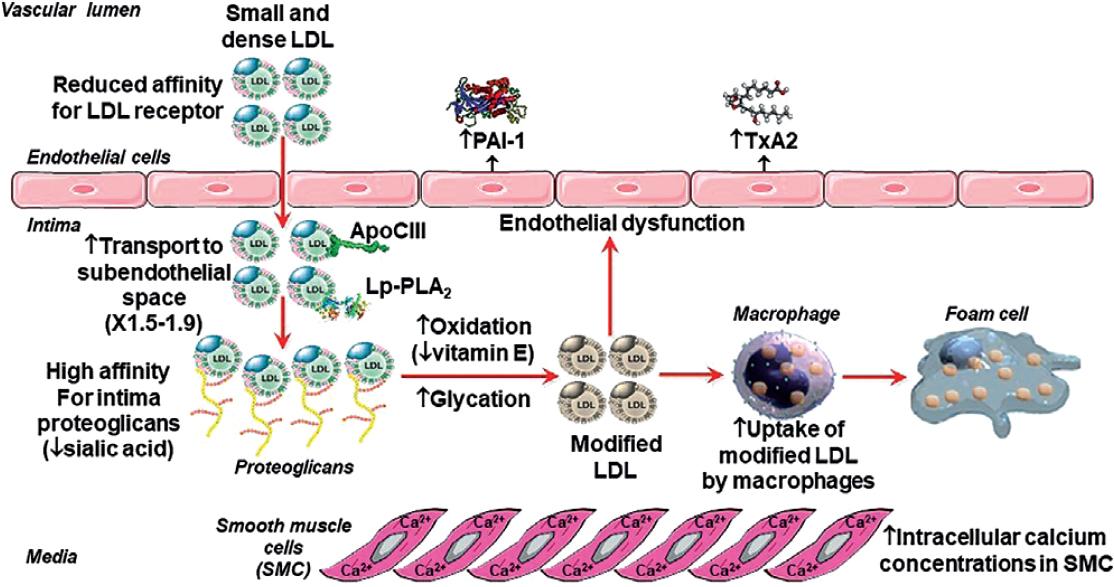
Figure 2 Atherogenic properties of small dense low-density lipoproteins. PAI-1: plasminogen activator inhibitor type 1; TxA2: thromboxane A2.
The above-described peculiarities of sdLDL have clinical implications. The number of apoB-containing lipoproteins in the artery lumen is the primary determinant of the rate at which these particles enter the arterial wall and remain trapped within the subendothelial space20-22. SdLDLs, containing less cholesterol, will easily enter and be trapped compared to the larger LDLs, which contain more cholesterol. On the other hand, the larger LDLs carry more cholesterol that will be released in the injured artery wall, causing more tissue damage23. Therefore, both large and small LDLs contribute to atherogenesis but mechanisms may differ between LDL classes. A systematic review of 24 studies that evaluated the relationships between LDL subfractions and cardiovascular events concluded that the number of LDL particles, rather than LDL size, is the main determinant of cardiovascular events rate24. Another clinical implication is the need for having an approach to identify cases with predominance of sdLDLs. The plasma accumulation of sdLDLs has a smaller than expected impact on LDL-cholesterol concentration. SdLDLs typically predominate in patients with moderately elevated triglycerides concentrations (i.e., type 2 diabetes and the metabolic syndrome). In these conditions, the ability of LDL-cholesterol to assess the atherogenic burden is not optimal. Alternatives are the non-HDL cholesterol and apolipoprotein B concentrations. The former assumes that all cholesterol, besides that transported in HDLs, is atherogenic. The latter is the best option because apoB level is a marker of the number of atherogenic lipoproteins. Consequently, in cases with plasma TRLs accumulation, clinicians should consider the use of apoB (if available) or non-HDL cholesterol to evaluate the atherogenic burden.
Plasma accumulation of other atherogenic particles (VLDLs and remnants) besides LDLs
VLDL particles and remnants have the same atherogenic risk as LDL25. In a Mendelian randomization analysis involving 654,783 participants, genetic variants of the LPL gene associated with lower triglycerides' concentrations and LDL-C-lowering variants of the LDLR gene were associated with a similarly lower risk of coronary heart disease per each 10 mg/dL decrease of apoB levels (odds ratios of 0.771 and 0.773, respectively)26. The benefit of reducing TRLs is similar to that obtained by decreasing LDL-C if adjusted per unit of change in apoB levels. Because LDL has a longer half-life in plasma, in physiologic conditions there are much more LDL particles than TRLs in plasma. In patients with normal triglyceride levels, for each VLDL particle, there are approximately nine LDL particles. In hypertriglyceridemia, the proportion is remarkably smaller; the extreme scenario is untreated dysbetalipoproteinemia, in which plasma abundance is greater for remnants than LDLs. A recent Danish report showed that the hazard risk for having cardiovascular outcomes is greater for VLDL than LDL (assessed by the number of particles measured by nuclear magnetic resonance spectroscopy). The hazard ratio (HR) for myocardial infarction (MI) was 3.5 times for VLDL, and 1.3 times for IDL and LDL combined. The authors stated, “VLDL particles are more atherogenic than LDL particles and VLDL and LDL should be measured separately”27.
There are several possible explanations for the greater atherogenic potential of VLDL compared to LDL. Remnants carry more cholesterol per particle than smaller LDLs. Indeed, a typical remnant in the IDL range may contain a fourfold greater absolute number of cholesterol molecules (up to ≈ 8600 per particle) as compared to an LDL particle (2000-2700). In addition, these particles more easily are trapped in the intima of the arterial wall, due to its apoC-III enrichment. TRLs plasma accumulation is associated with increased C-reactive protein (CRP), a marker of low-grade inflammation. As a result, TRLs by themselves can promote intimal inflammation, plaque rupture, and MI.
The greater susceptibility of remnants to suffer chemical modifications increases the atherogenicity of these particles at the same extent that modified LDLs. Both types of particles affect in a similar magnitude the expression of adhesion molecules on monocytes and endothelium and the expression of inflammatory genes in vascular cells through a redox-sensitive mechanism. Both are chemotactic for monocytes, T cells and tissue macrophages and induce the formation of macrophage foam cells. Both alter endothelial cell relaxation by decreasing NO activity. Finally, chemically modified remnants and oxidized LDLs have similar proatherogenic effects on endothelial cells and smooth muscle cells (i.e., cytotoxicity and induction of apoptosis, stimulation of the expression of tissue factor, and facilitation of platelet aggregation). Finally, a mechanism by which modified TRLs, but not LDLs, may contribute to atherogenesis is the activation of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) in endothelial cells, which makes them dysfunctional28.
TRLs induce endothelial dysfunction and low-grade inflammation of the vascular wall, and are retained in the subendothelial space and the atherosclerotic plaques
TRLs alter endothelial function through several mechanisms29, schematized in figure 3. Remnants affect the autophosphorylation of focal adhesion kinase and its downstream signaling pathway, phosphatidylinositol 3-kinase/protein kinase B (Akt), causing the inactivation of NO synthase (NOS). This results in a decrease in the endothelial synthesis of NO (eNOS). Moreover, plasma TRLs accumulation is associated with higher asymmetric dimethyl arginine, an endogenous inhibitor of eNOS30. In addition, TRLs induce endothelin-1 (ET-1) release in humans31. ET-1 induces vasoconstriction by increasing the tone of vascular smooth muscle cells, increases the proliferation of these cells, promoting thrombosis, inflammation, and eNOS uncoupling32. When eNOS becomes uncoupled, it generates the highly oxidizing species superoxide instead of NO. ET-1 stimulates the expression and activity of arginase-2 in endothelial cells and the activity of this enzyme in macrophages. This favors formation of reactive oxygen species (ROS). Arginase is a critical reciprocal regulator of NO production by competing with eNOS for the substrate L-arginine in endothelial cells, thereby reducing the bioavailability of NO.
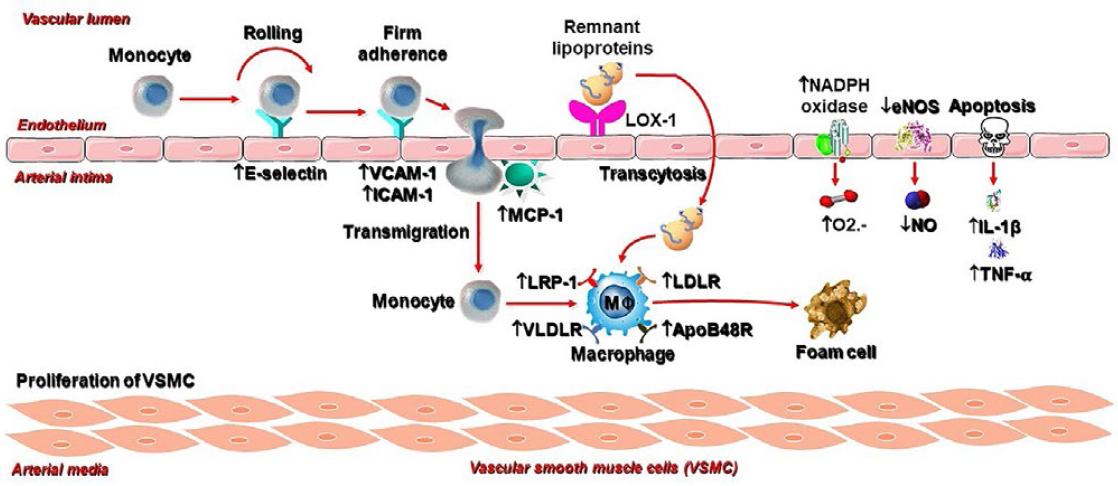
Figure 3 Pro-inflammatory and pro-atherogenic actions of remnant particles. LOX-1: lectin-like oxidized low-density lipoprotein (LDL) receptor-1; VCAM-1: vascular cell adhesion molecule-1; ICAM-1: intercellular adhesion molecule-1; MCP-1: monocyte chemoattractant protein-1; LRP-1: LDL receptor related protein-1; VLDLR: VLDL receptor; LDLR: LDL receptor; apoB48R: apob48 receptor; O2–: anion superoxide; eNOS: endothelial nitric oxide (NO) synthase; NADPH: nicotinamide adenine dinucleotide phosphate; IL-1β: interleukin-1 β; TN-α: tumor necrosis factor -α.
On the other hand, remnants induce endothelial cell apoptosis via increased secretion of pro-apoptotic cytokines. In addition, neutral and oxidized free fatty acids decrease eNOS activity, increase endothelial permeability, and increase ROS production by endothelial cells by activating the enzymes NADPH oxidase and cytochrome P450 2C933.
TRLs cause low-grade inflammation at the arterial wall by direct and indirect mechanisms34. These lipoproteins stimulate the endothelial expression of molecules that mediate the adhesion of circulating monocytes to the endothelial surface. Among them are ICAM-1 and VCAM-1. Likewise, TRLs increase the endothelial expression of Monocyte chemoattractant protein-1, which facilitate the trans-endothelial migration of monocytes toward the subendothelial space. Among TRLs, remnants are the main contributor; exposure of endothelial cells to remnants induce superoxide production by the enzyme nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) and the secretion of inflammatory cytokines (i.e., interleukin 1β and tumor necrosis factor-α) through the activation of the LOX-1 receptor. Another mechanism by which TRLs induce inflammation is the activation of the inflammasome nucleotide-binding domain-like receptor family pyrin domain-containing protein 1 (NLRP1)35. The precise mechanism of the inflammasome activation remains to be identified, although the local release of fatty acids from the VLDL triglycerides may be involved.
LPL, an enzyme located at the endothelial surface or within the arterial intima, hydrolyzes TRLs-triglycerides into free fatty acids and monoacylglycerols, which generate local inflammation, especially oxidized free fatty acids. These end products potentiate the inflammatory stimulus above described. Furthermore, endothelial cell apoptosis and greater permeability of the endothelial monolayer are induced36. On the other hand, remnant cholesterol in the subendothelial space induces macrophage M1 polarization and potentiates inflammation. Pro-inflammatory macrophages secrete a spectrum of factors, which promote lesion development and ultimately destabilization; these include matrix-degrading proteases, pro-oxidant enzymes, lipases, growth factors, pro-inflammatory cytokines, eicosanoids, proteoglycans, and procoagulant factors.
Several mechanisms of entry of lipoproteins to the subendothelial space participate in the atherosclerotic process37. Transcytosis is a transport system in which lipoproteins and other macromolecules are transported through endothelial cells through specialized clathrin-coated vesicles. These particles have a diameter of approximately 100 nm. Therefore, lipoproteins larger than 75-80 nm (i.e., chylomicrons and larger VLDLs) cannot enter the arterial intima through transcytosis and an intact endothelium. This size limitation explains that individuals with chylomicronemia or familial hypertriglyceridemia are not associated with premature or severe atherosclerosis. The smaller the lipoproteins, the higher their rate of entry into the arterial wall and greater the likelihood of atherosclerotic lesions. The capacity of the transport system by transcytosis is very high. It has been estimated that approximately 2500 transport vesicles leave the plasma membrane every minute. Therefore, it is not the influx of lipoproteins, but rather the selective accumulation of particles in the arterial wall. Retention is mediated by ionic interactions between positively charged amino acid residues (arginine and lysine) in apoB and apoE of atherogenic lipoproteins with negatively charged sugar and sulfate groups in glycosaminoglycan chains of subendothelial proteoglycans. Once trapped, LDLs undergo chemical modification (e.g., oxidative modification) that increases their recognition by the macrophage's scavenger receptors, leading to the formation of foam cells7. In contrast, remnants and the smaller VLDLs can be taken up by macrophages without oxidative modification. TRLs are taken up by macrophages by receptor-mediated and non-receptor-mediated routes. Uptake through either the LDLR, the LDLR-related protein 1, or the VLDL receptor is apoE-dependent. Furthermore, uptake could be mediated by the apoB48 receptor (apoB48R); this receptor recognizes apoB48 and the apoB48 section or portion of apoB100 of TLRs38. Other minor uptake mechanisms are the CD36 scavenger receptor and the SR-B1 receptor38. Confirmatory evidence of the participation of TRLs in atherogenesis is the demonstration of ApoB48 and apoB100 in human aortic atherosclerotic plaques39.
TRLs increase plasma viscosity and favor a procoagulant state
Plasma TRLs accumulation causes an exponential increase in plasma viscosity40. This feature is independent of the associated increment of fibrinogen and protein levels, which are other major determinants of plasma viscosity. High blood viscosity can lead to atherothrombosis by producing alterations in microcirculatory blood flow, damage by sheer stress at the blood-endothelium interface and an increase in the propensity for thrombosis. Furthermore, TRLs are associated with increased platelet aggregation and higher concentrations of blood factors I (fibrinogen), VII, VIII, and X and tissue factor. Finally, TRLs cause upregulation of PAI-1 and its antigen and decrease of the activity of tissue-type plasminogen activator.
Comparison of the atherogenic burden caused by TRLs and LDLs
Overall, the population attributable risk is greater for LDLs because the number of LDL particles is remarkably higher than the TRLs in the plasma, even in the postprandial periods. The ratio of LDL/TRL particles depends on the plasma triglyceride concentration. If plasma triglycerides are below 133 mg/dL, the ratio LDL to TRLs is 9 to 1. However, the ratio is significantly lower if triglycerides are above 265 mg/dL (4 to 1)41. Even so, this concept is in transition. Due to their larger size, TRLs transport 5-20 times more cholesterol per particle than LDL. In the Copenhagen General Population Study (n = 9293), authors estimated, using nuclear magnetic resonance spectroscopy, that one-third (32%) of the plasma cholesterol pool in non-fasting plasma is transported in remnants, one-third (35%) in LDLs, and the remaining third (32%) in HDLs. In a recent publication derived from the same study (including 1816 cases of MI with a mean follow-up of 11 years), VLDL cholesterol accounted for half the risk of MI associated with plasma lipids, while VLDL triglycerides did not explain the risk42. Importantly, 50% of the cholesterol found in atherosclerotic plaque comes from TRLs cholesterol43.
Clinical and therapeutic approach of cases with TRLs plasma accumulation
The hallmark to identify subjects with plasma accumulation of atherogenic TRLs is mixed hyperlipidemia (defined as cholesterol above 200 mg/dL and triglycerides above 150 mg/dL). Concentrations above 150 mg/dL should alert physicians about TRLs accumulation as a contributing factor for increased cardiovascular risk. The concomitant increment of cholesterol is a marker of the presence of the atherogenic TRLs subclasses (i.e., small VLDLs, IDLs, and remnants). The best example is dysbetalipoproteinemia, in which plasma accumulation of remnants and IDLs is the explanation of the abnormal lipid profile. This atherogenic primary dyslipidemia causes moderate to severe mixed hyperlipidemia, in which the triglycerides and cholesterol concentrations are very similar (i.e., around 300 mg/dL).
An etiologic diagnosis is the next step in the evaluation of cases with plasma TRLs accumulation. The most common diseases associated with accumulation of atherogenic TRLs are familial combined hyperlipidemia, type 2 diabetes (and its preceding conditions), and lifestyle habits or medications that cause decreased insulin action. On the other hand, primary and polygenic chylomicronemias, familial hypertriglyceridemia and excessive intake of alcohol, simple sugars, saturated and unsaturated fats are the conditions to be searched in cases with isolated hypertriglyceridemia (moderate to severe hypertriglyceridemia with normal cholesterol concentrations (or a ratio triglycerides/cholesterol ≥ 5). These conditions are associated with plasma accumulation of non-atherogenic TRLs (i.e., chylomicrons and/or VLDL1).
TRLs plasma accumulation interferes with the assessment of LDL-cholesterol related risk. The LDL-cholesterol estimation is under-appreciated when applying the most used formula (i.e., Friedewald equation). Potential solutions are the estimation of the cholesterol transported in LDLs using other formulas (Martin or Sampson equations)44. A second option is to assume that all cholesterol besides HDL-cholesterol is potentially atherogenic. For this purpose, the non-HDL cholesterol is calculated (Non-HDL cholesterol = cholesterol – (HDL-cholesterol). However, these options do not take into account the abnormal composition of the sdLDLs. The best approach is to estimate the number of potential atherogenic particles in the plasma. The most convenient option is to measure the apoB plasma concentrations. However, its cost (i.e., similar to a complete lipid profile) and poor access in clinical laboratories limit its routine use. Another option is the NMR-spectroscopy of plasma lipoproteins, in which the number of lipoprotein particles classified by subclasses is informed. This method is costly and is not available in the majority of the clinical laboratories. The lipid guideline by the European Atherosclerosis Society recommends the apoB measurement in high-risk cases (i.e., type 2 diabetes and primary dyslipidemias) in which TRLs excess is suspected45. Regrettably, it is not possible to measure remnant and other TRLs in the clinical laboratories. Although a formula has been proposed to estimate remnant cholesterol (Remnant cholesterol = Cholesterol – HDL cholesterol – LDL cholesterol), this approach is affected by the methodologic limitations of the LDL-cholesterol estimation. A colorimetric method is currently under evaluation, and more studies are required for its inclusion in clinical practice.
The persistence of atherogenic TRLs is a common explanation for the residual risk of statin-treated cases. This concept integrates the cardiovascular events that occur even if LDL-cholesterol treatment goals are reached and sustained. Looking for new alternatives to correct TRLs excess is one of the most active areas of research in the cardiovascular prevention field43. It implies new therapeutic goals and innovative drug therapies. ApoB levels may become, in the near future, the main treatment target because it integrates the LDL– and TRL-related atherogenic burden. According to the European Society of Cardiology/European Atherosclerosis Society (ESC/EAS) guidelines, the current proposed treatment targets are <80 mg/dL plus a 50% reduction in the basal concentration in high-risk cases (primary prevention with severe risk factors). The corresponding values for very high-risk subjects are <65 mg/dL (secondary prevention and/or primary prevention with various severe risk factors)45. However, these thresholds should be confirmed using meta-analyses of clinical trials. Another approach is to consider the accomplishment of both LDL-cholesterol and non-HDL cholesterol treatment targets. Currently, the primer goal is to decrease LDL- cholesterol; non-HDL cholesterol targets are considered as a secondary goal, especially in patients with type 2 diabetes. The accepted non-HDL cholesterol goals are < 100 mg/dL plus a 50% reduction in the basal concentration in high-risk cases (primary prevention with severe risk factors). The corresponding values for very high-risk subjects are < 85 mg/dL (secondary prevention and/or primary prevention with several severe risk factors).
Beyond current options (fibrates and omega-3 fatty acids), innovative therapies for TRL are under intensive evaluation. The most promising alternative is the activation of the LPL pathway, by decreasing the activity or concentration of natural inhibitors of the enzyme (i.e., apolipoprotein C-III, and angiopoietin-like protein 3 [ANGPTL3]), using monoclonal antibodies or antisense drugs (reviewed in detail in43 (i.e., volanesorsen and akcea-apoC-III vs. apoC-III and evinacumab, vupanorsen and aro-ANG3 vs. ANGPTL3). Both, apoC-III– and ANGPTL3-targeted therapies, lower remnants and TRLs concentrations. Furthermore, these therapies have a positive effect on triglycerides (30-60% decrement), LDL-cholesterol (5-30% decrement), and apoB (4-25% decrement). Additional studies are needed to evaluate their ability to reduce cardiovascular risk, especially in high-risk cases with remnant-related dyslipidemias.
A current alternative is the selective activation of the peroxisome proliferator-activated receptor alpha (PPAR alpha). These nuclear receptors activate or suppress the expression of multiple key genes involved in lipid metabolism. Fibrates, which are PPAR alpha agonists, are currently an option for drug therapy for hypertriglyceridemia. Fibrate therapy decreases remnants and other TRLs. However, clinical trials in which fibrates had been used to reduce the incidence of cardiovascular events had found a marginal effect; methodologic deficiencies contributed to the non-satisfactory results. New agents, like pemafibrate, were developed to increase its selectivity for the promoter regions of the genes related with lipoprotein metabolism. This agent seems to be safer and slightly more effective for reducing plasma triglycerides and apoB levels than fenofibrate. The PROMINENT trial will assess the effect of pemafibrate to reduce the incidence of cardiovascular events in patients with type 2 diabetes46.
Highly purified eicosapentanoic acid (EPA), alone or in combination with other omega-3 fatty acids, has been used to treat hypertriglyceridemia. Special interest was raised by the REDUCE-IT trial, in which 4g/day EPA diminished in 25% the relative risk of cardiovascular events in high-risk patients on background statin therapy. However, a secondary analysis showed that the potential benefit is related with EPA concentrations, not with changes in triglycerides or remnants levels. In the STRENGHT trial, 4 g/day of EPA/DHA did not reduce the relative risk of cardiovascular events. Up to now such discordance is explained by important differences between EPA and DHA structures and membrane actions. Thus, either EPA or other omega-3 products may not be considered as options to prevent coronary events through increasing remnants removal from plasma47.
Finally, the relationship of remnant cholesterol levels with the progression of atherosclerotic disease during lipid lowering therapy is of special interest. The contribution of remnant cholesterol to changes in percent coronary atheroma volume (determined by intravascular ultrasound) and 2-year major cardiovascular events is greater than the observed for LDL-cholesterol in ten clinical trials (n = 5754) involving both statin and non-statin therapies48.
CONCLUSIONS
As summarized in figure 1, atherogenic TRLs are a potential target for the prevention and treatment of atherosclerosis. TRL cholesterol accumulates in the plaques, and TRLs triglycerides exacerbate the inflammatory component of the disease. Physicians should be able to distinguish cases with plasma accumulation of atherogenic TRLs (by the presence of mixed hyperlipidemia, high non-HDL cholesterol or apoB levels) from other causes of hypertriglyceridemia. Upcoming evidence of ongoing clinical trials will have a major impact on the current clinical guidelines putting remnants and other atherogenic TRLs as a major research area and a complimentary objective, besides LDL cholesterol, for cardiovascular prevention.

