











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkMódulo 1: Clasificación de gammapatías monoclonales
Las gammapatías monoclonales son un grupo de patologías derivadas de células plasmáticas poscentro germinal que han recuperado su capacidad de replicación, esta serie de condiciones no son obligatoriamente secuenciales1. Su espectro clínico abarca desde la presencia asintomática de una proteína monoclonal en suero u orina hasta estados de enfermedad con destrucción de tejido sano. Los síntomas pueden ser resultado de la infiltración de las células plasmáticas clonales y de la proteína monoclonal secretada o de la interacción de la célula plasmática con su microambiente2.
Las gammapatías monoclonales, sus criterios diagnósticos y tasa de progresión se engloban en la tabla 1.
Tabla 1 Criterios diagnósticos y tasa de progresión de las gammapatías monoclonales
| Patologías | Criterios diagnósticos | Progresión |
|---|---|---|
| MGUS no IgM | - Proteína monoclonal sérica < 3 g/dl - Células plasmáticas clonales < 3% - Ausencia de daño a órgano blanco |
1%/año |
| Mieloma tipo smoldering | - Proteína monoclonal sérica > 3 g/dl o proteína
monoclonal en orina > 500 mg/24 horas y/o células plasmáticas
clonales 10-60% - Ausencia de eventos definitorios de mieloma o amiloidosis* |
10%/año |
| MGUS IgM | - Proteína monoclonal sérica IgM < 3g/dl - Infiltración linfoplasmocítica en médula ósea < 10% - Ausencia de daño a órgano blanco |
1-5%/año |
| MGUS cadenas ligeras | - Radio cadenas ligeras < 0.26 o > 1.65 - Incremento de la cadena ligera involucrada (kappa relación > 1.65 y lambda < 0.26) - No existe expresión de cadena pesada en inmunofijación - Ausencia de daño a órgano blanco - Células plasmáticas clonales < 10% - Proteína monoclonal en orina < 500 mg/24 horas |
0-3%/año |
| Plasmocitoma solitario | - Biopsia de una lesión ósea o tejido blando con
evidencia de células plasmáticas clonales - Médula ósea sin evidencia de infiltración por células plasmáticas - Hallazgos esqueléticos normales - Ausencia de daño a órgano blanco |
10%/3 años |
| Plasmocitoma solitario con involucro a médula ósea | - Biopsia de una lesión ósea o en tejido blando con
evidencia de células plasmáticas clonales - Médula ósea con < 10% de células plasmáticas - Resonancia magnética o tomografía de columna o pelvis sin lesiones - Ausencia de daño a órgano blanco |
60%/3 años (médula ósea) 20%/3 años (tejido blando) |
*Hipercalcemia mayor de 11 mg/dl, insuficiencia renal (depuración de creatinina menor de 40 ml/min, creatinina sérica mayor de 2 mg/dl), anemia menor de 10 g/dl, lesiones líticas detectadas por radiografía, tomografía o tomografía por emisión de positrones-tomografía computarizada.
MGUS: gammapatía monoclonal de significado incierto; IgM: inmunoglobulina M.
Existen otras entidades relacionadas con gammapatía monoclonal, las cuales se enlistan en la tabla 2.
Tabla 2 Otras entidades relacionadas con gammapatía monoclonal
| Causas de proteína monoclonal en otras enfermedades | |
|---|---|
| Asociadas a diferentes patologías |
Hematológicas
- Síndromes linfoproliferativos (linfomas, leucemia linfocítica crónica o de células peludas) - Síndromes mielodisplásicos-mieloproliferativos (leucemia mieloide crónica, policitemia vera, mielofibrosis) - Hemoglobinuria paroxística nocturna, aplasia pura de serie roja |
|
Autoinmunes e inmunosupresión
- Lupus eritematoso sistémico, artritis reumatoide - Esclerodermia, polimiositis, tiroiditis - Trasplante de órgano sólido | |
|
Dermatológicas
Esclerodermia, pioderma gangrenoso, xantulograma | |
|
Virales
- Virus de hepatitis C y virus de la inmunodeficiencia humana | |
|
Endocrinológicas
- Hiperparatiroidismo | |
|
Misceláneas
Hemosiderosis pulmonar, sarcoidosis y neoplasias | |
| Asociadas a proliferación linfoplasmocitaria | - Amiloidosis - Enfermedad de cadenas pesadas o ligeras - Macroglobulinemia/linfoma linfoplasmocítico - Plasmocitoma o mieloma múltiple (solitario, extramedular o síndrome de POEMS) |
POEMS: polirradiculoneuropatía (P), organomegalia (O), endocrinopatía (E), trastorno de proliferación clonal de células plasmáticas (M) y cambios cutáneos (S).
Gammapatía monoclonal de significado incierto
La gammapatía monoclonal de significado incierto (MGUS) se considera el primer paso previo al mieloma múltiple (MM), puede estar presente de los 8 a 10 años previos su diagnóstico. Sin embargo, hasta el 25% de los pacientes con MM nunca tuvieron o no se identificó esta alteración3.
Su incidencia aumenta con la edad, estimándose desde el 3 hasta el 20% en mayores de 50 años, y es mayor en hombres y sujetos de raza negra. Se estima que el riesgo de progresión a MM es del 1% por año4.
El proceso de conversión conlleva múltiples pasos, algunos de los cuales se llevan a cabo de manera simultánea. Inicialmente la célula plasmática poscentro germinal debe sufrir alteraciones cromosómicas, principalmente hiperdiploidía, así como traslocaciones en la cadena pesada de la inmunoglobulina (IgH), esto tiene como consecuencia la activación del oncogén KRAS, inactivación del TP53 e inestabilidad genómica. A la par, se presenta rearreglo en el microambiente celular; como consecuencia, la célula plasmática recobra su capacidad de proliferación, lo que conlleva la monoclonalidad de la patología5.
Criterios pronósticos
Existen actualmente dos modelos de predicción de riesgo, desarrollados por la Mayo Clinic y por el grupo PHETEMA (Programa español de tratamientos en hematología), que toman en cuenta los elementos que se muestran en la tabla 3.
Tabla 3 Criterios de riesgo MGUS
| Criterios de riesgo MGUS | Seguimiento | |||
|---|---|---|---|---|
| Factores de riesgo | Riesgo | Número de factores a 20 años | Riesgo de progresión | |
| Modelo Mayo Clinic | ||||
| - Isotipo no IgG - Proteína M > 1.5 g/d - Rel. CL alterada |
Bajo | 0 | 5% | Valoración cada 6 meses por 2 años, posteriormente cada año |
| Intermedio | 1 | 21% | Lo anterior más aspirado de médula ósea; si incrementa paraproteína > 25% descartar amiloidosis y CRAB | |
| 2 | 37% | |||
| Alto | 3 | 58% | ||
| Modelo PETHEMA | A 5 años | |||
| Fenotipo anormal Aneuploidía | 0 | 2% | ||
| 1 | 10% | |||
| 2 | 46% | |||
MGUS: gammapatía monoclonal de significado incierto; IgG: inmunoglobulina G; CRAB: hipercalcemia, insuficiencia renal, anemia y lesiones líticas; PHETEMA: Programa español de tratamientos en hematología; Rel. CL : Relación de Cadenas Ligeras.
Adaptada de Maciocia, et al., 20172.
Mieloma múltiple quiescente o asintomático
Su incidencia es desconocida en nuestro país. En EE.UU. se reportan 0.9 casos por cada 100,000 habitantes, de estos el 13.7% evoluciona a MM sintomático en mayores de 67 años6.
Criterios diagnósticos y pronósticos (Tabla 4)
Tabla 4 Criterios de riesgo mieloma quiescente
| Criterios de riesgo mieloma qsuiescente | Seguimiento | ||||
|---|---|---|---|---|---|
| Factores de riesgo | Número de factores | Riesgo de progresión a 5 años | Mediana TTPa | ||
| Modelo Mayo Clinic | |||||
| - Rel. CL anormal - CP en MO > 10% - Proteína M > 3.0 g/dl |
Riesgo bajo | 1 | 25% | 10 años | Valoración anual si no incrementa paraproteína >25% |
| Riesgo intermedio | 2 | 51% | 5 años | Valoración trimestral durante el primer año descartando CRAB | |
| Riesgo alto | 3 | 76% | 1.9 años | Valoración bimensual, considerar tratamiento precoz | |
| Modelo PETHEMA | A 5 años | ||||
| - > 95% CP anormales por citometría de
flujo - Inmunoparesis |
0 | 4% | NA | ||
| 1 | 46% | 6 años | |||
| 2 | 72% | 1.9 años | |||
TTPa: Tiempo Parcial de tromboplastina activada; Rel.CL: Relación de cadenas ligeras; CP: Celúlas plasmáticas ;MO: médula ósea; NA: No aplica; HETEMA: Programa español de tratamientos en hematología; CRAB: hipercalcemia, insuficiencia renal, anemia y lesiones líticas.
Adaptada de Maciocia, et al., 20172
Los criterios para que un paciente sea considerado de ultra alto riesgo son los siguientes:
Estos criterios aumentan el riesgo de progresión a más del 70% a dos años.
Plasmocitoma solitario
El plasmocitoma solitario equivale al 2-5% de las gammapatías monoclonales. Se caracteriza por la proliferación extramedular de células plasmáticas en ausencia de monoclonalidad en medula ósea u otros datos de daño a órgano blanco (hipercalcemia, insuficiencia renal, anemia y lesiones líticas [CRAB])9.
La sintomatología depende de la destrucción del tejido afectado por el tumor, el Plasmocitoma solitario óseo (PSO) se presenta con mayor frecuencia en esqueleto axial y el plasmocitoma solitario extramedular (PSE) se presenta mayormente en nasofaringe y orofaringe. Este tipo de tumor no tiene alteraciones patognomónicas por imagen, por lo que para integrar el diagnóstico definitivo se requiere de la demostración de infiltración de células plasmáticas monoclonales obtenidas mediante biopsia10.
Criterios pronósticos
‒ Localización y tamaño del tumor: la presentación de PSO tiene una sobrevida libre de mieloma menor que la que confiere el PSE (36.4 vs. 72.1% a 10 años). Un tumor mayor de 5 cm confiere un pronóstico desfavorable11.
‒ Paraproteína y cadenas ligeras: la persistencia de proteína monoclonal posterior a la radioterapia es un factor de riesgo relacionado con progresión a MM11.
Tratamiento
La radiación es actualmente el tratamiento de elección. La dosis que administrar puede ser de 40-50 Gy, variable11. El uso de cirugía y quimioterapia es más controvertido que la medida anterior.
Síndrome de POEMS
El síndrome de POEMS (polirradiculoneuropatía [P], organomegalia [O], endocrinopatía [E], trastorno de proliferación clonal de células plasmáticas [M] y cambios cutáneos [S]) es un síndrome paraneoplásico que se presenta con una discrasia de células plasmáticas. Su nombre deriva del acrónimo acuñado por el Dr. Bardwick en 1980, sin embargo, este síndrome no se limita a las alteraciones que componen el acrónimo; los criterios diagnósticos se han modificado para dar lugar a una clasificación más amplia, ya que también se presentan con igual frecuencia: papiledema, sobrecarga extravascular, lesiones óseas escleróticas y trombocitosis/eritrocitosis1.
La fisiopatología de esta entidad es poco conocida y se sabe de la participación del factor de crecimiento endotelial vascular (VEGF), sin embargo, se debe diferenciar de las gammapatías ya descritas debido a las variaciones en el manejo y pronóstico. Se cuenta con criterios diagnósticos establecidos para diferenciar de otras discrasias sanguíneas (Tabla 1)2. Su epidemiologia está mal determinada debido al escaso número de casos con que se cuenta incluso en centros de referencia; se estima que en Japón, uno de los países con mayor índice de casos reportados, se presentan 0.3 casos por cada 100,000 habitantes3.
Tabla 1 Criterios diagnósticos para síndrome de POEMS
| Criterios diagnósticos para síndrome de POEMS | |
|---|---|
| Criterios mayores mandatorios | 1. Polineuropatía desmielinizante |
| 2. Discrasia monoclonal de células plasmáticas | |
| Otros criterios mayores | 3. Enfermedad de Castleman |
| 4. Lesiones óseas escleróticas | |
| 5. Elevación sérica del VEGF | |
| Criterios menores | 6. Organomegalia |
| 7. Sobrecarga hídrica extravascular | |
| 8. Endocrinopatía | |
| 9. Cambios en la piel (hiperpigmentación, hipertricosis, plétora) | |
| 10. Papiledema | |
| 11. Trombocitosis/policitemia | |
| Otros síntomas | 12. Pérdida de peso, hiperhidrosis, hipertensión pulmonar, diarrea |
POEMS: polirradiculoneuropatía (P), organomegalia (O), endocrinopatía (E), trastorno de proliferación clonal de células plasmáticas (M) y cambios cutáneos (S); VEGF: factor de crecimiento endotelial vascular.
Adaptada de Dispenzieri, 20171.
El diagnóstico requiere de una gran cantidad de estudios para determinar el grado de daño o afectación de cada órgano o sistema que se sabe que puede ser afectado por el síndrome. Una historia clínica completa, incluyendo exploración física neurológica completa e interrogatorio poniendo énfasis en alteraciones de la menstruación, en mujeres, o en otras alteraciones de probable origen endocrinológico, es de suma importancia. En segundo lugar, los estudios de imagen y gabinete deben realizarse orientados por los hallazgos clínicos y del interrogatorio, de manera que podamos determinar los criterios cumplidos con la menor cantidad de recurso utilizado.
De la totalidad de síntomas y signos que pueden presentarse, los más frecuentes son: neuropatía (en hasta el 100% de los casos), edema periférico, hiperpigmentación de la piel, trombocitosis y discrasia de células plasmáticas (estos últimos en hasta el 88% de los casos). No existen modelos predictores de pronóstico específicos para esta entidad, pero se ha demostrado que su supervivencia, tanto libre de progresión como global, es mejor que la de otras gammapatías (supervivencia global [SG] a 10 años de hasta un 79%)4.
El tratamiento del síndrome de POEMS se determinará con base en el grado de infiltración de células plasmáticas a medula ósea. En aquellos casos en que la infiltración sea escasa o nula, la radioterapia se considera el tratamiento estándar, con tasas de respuesta del 50 al 70%, con acusada mejoría de la sintomatología. Por otra parte, cuando se tiene un importante componente de discrasia de células plasmáticas, el tratamiento de elección debe estar inspirado en aquellos utilizados en este grupo de enfermedades. No existe un tratamiento estándar y se han reportado resultados no concluyentes con diversos esquemas de tratamiento, tomando como base el uso de inhibidores del proteosoma (IP) en combinación con inmunomoduladores (IMiD), como talidomida y lenalidomida, con buenas tasas de respuesta6. Sin embargo, el trasplante de progenitores hematopoyéticos sigue reportando tasas de respuesta de hasta el 100%, con supervivencia libre de enfermedad del 72% a seis años en una serie de la Mayo Clinic4.
El seguimiento y la determinación de la respuesta se hace de acuerdo con los órganos afectados de manera inicial y al tratamiento administrado; así, si se decide utilizar IP más IMiD, el tiempo de valoración de la respuesta debe ser a los cuatro meses de manera inicial, para posteriormente ser revalorado a los seis meses (Tabla 2).
Tabla 2 Criterios de respuesta
| Criterios de respuesta | |||
|---|---|---|---|
| Parámetro | RC | Mejoría | Progresión |
| VEGF plasmático | Normal | 50% de reducción de basal | 50% de aumento de basal |
| Hematológico | Inmunofijación negativa en suero y orina | 50% de reducción del pico monoclonal | 25% de aumento del valor basal |
| Ascitis/edema | Ausente | Mejoría en 1 punto por CTCAE | Empeoramiento por 1 punto de CTCAE |
| Presión sistólica vent. por Eco TT | Normal | Menor 40 mmHg | NA |
| Papiledema | Ausente | Ausente | Presente |
RC: respuesta completa; VEGF: factor de crecimiento endotelial vascular; CTCAE: Common Terminology Criteria for adverse Events; NA: No aplica; vent.: Ventricular; Eco TT: Ecocardiografia Transtoracica.
Adaptada de Dispenzieri, 20172.
Enfermedad de Castleman y síndrome de POEMS
La enfermedad de Castleman fue descrita originalmente en la década de los 50, específicamente en el caso de un paciente con nódulos mediastinales, fiebre y pérdida de peso en quien se sospechaba de diversas patologías, entre ellas tuberculosis, las cuales fueron descartadas posteriormente a la descripción de la infiltración plasmocítica que se encontró en dichos ganglios, correspondiendo a lo que ahora conocemos como enfermedad de Castleman unicéntrica5.
Esta enfermedad se puede encontrar relacionada a diversas patologías tanto infecciosas (virus de la inmunodeficiencia humana [VIH], herpesvirus humano 8) como autoinmunes y, en el caso que nos atañe, hematológicas. Si bien se la considera una enfermedad benigna, confiere un aumento en el riesgo de linfoma y otras neoplasias.
En lo que respecta a su fisiopatología, esta dependerá del tipo de enfermedad que estemos enfrentando. En ambos casos existe el aumento de interleucina (IL) 6, con la sobreproliferación linfocítica que da lugar al aumento de factor estimulante de VEGF y a la enfermedad hialino-vascular, la diferencia radica en la relación de los virus previamente mencionados para la multicéntrica y la casi exclusiva actividad de la IL-6 para la presentación unicéntrica. Se puede encontrar una variante de Castleman en la cual hay proliferación e infiltración de células plasmocíticas con sobreexpresión de inmunoglobulina (Ig) G o IgA y restricción de cadenas lambda. Es esta variante la que debe considerarse al momento de hacer un diagnóstico diferencial entre síndrome de POEMS y macroglobulinemia de Waldenström (MW). Esta forma plasmocítica se ha reportado en hasta el 44% de los casos de Castleman multicéntrico6.
El diagnóstico es histopatológico y de exclusión. El abordaje de pacientes en quienes se sospeche de Castleman o POEMS debe incluir básicamente los mismos estudios descritos en el síndrome anterior. Se debe llevar a cabo serología para descarte de VIH y sífilis. También se deben descartar enfermedades autoinmunes que puedan tener relación con la enfermedad. En el caso específico de la correlación entre Castleman y POEMS, esta se basa en el reporte histopatológico de la primera, con características clínicas del segundo, es así que varios casos de enfermedad de Castleman con características «singulares» en realidad corresponden a Castleman en el marco de POEMS. La neuropatía presente en el Castleman suele ser más leve que la presentada en POEMS y remite de manera parcial. En las pruebas de laboratorio se observa restricción de cadenas lambda, pero con niveles de IgM normales, tendiendo más a elevación policlonal de IgG e IgA, lo que ayuda a la diferenciación de MW y de mieloma múltiple (MM)1.
El tratamiento de la enfermedad de Castleman es variado, no se cuenta con tratamiento estándar y la gran mayoría de los esquemas utilizados están fuera de indicación real. En un metaanálisis publicado en 2016 se encontraron esquemas basados en esteroides, los cuales reportan supervivencia a dos años de hasta el 88%; otros basados en quimioterapia, como con ciclofosfamida, con supervivencias a dos años del 90%, y esquemas con uso de rituximab con supervivencias de hasta el 100% a dos años. Sin embargo, el bortezomib, los IMiD y otros quimioterapéuticos han sido descritos y utilizados con tasas de respuesta similares a las reportadas por los tratamientos más tradicionales. La IL-6 se ha convertido en el blanco de algunos tratamientos novedosos, específicamente para el tipo multicéntrico negativo a VIH, con tasas de respuesta variables6.
En el caso de relación documentada entre POEMS y Castleman, el tratamiento debe estar orientado a la resolución de la proliferación plasmocítica, con el uso de IP o inmunomodulares. Se sabe que esta relación, que se observa en entre el 15 y el 25% de los casos, tiene respuesta favorable a dichos tratamientos7.
La valoración de la respuesta y el seguimiento no varían en comparación a los descritos para síndrome de POEMS, el tratamiento de segunda línea dependerá de los recursos disponibles y de la respuesta a la primera línea de tratamiento.
Macroglobulinemia de Waldenström
Definición
De acuerdo con la última revisión de la Organización Mundial de la Salud en el 2016, la MW forma parte de las neoplasias de células B maduras, perteneciendo a un subconjunto de los linfomas linfoplasmocíticos. La MW es una entidad clinicopatológica definida por: infiltración de la médula ósea (MO) por linfoma linfoplasmocítico y la presencia de un componente monoclonal IgM en suero8.
Epidemiología y clasificación
La MW tiene una incidencia global de 3 por millón de personas por año y representa aproximadamente el 1-2% de los linfomas no Hodgkin9,10. La incidencia ajustada a edad es de 3.4 a 7.3 por millón en hombres, y de 1.7 a 4.2 por millón en mujeres, en la población de EE.UU. y Europa, respectivamente10,11. Contrario a la incidencia de MM, la prevalencia de la MW es mayor en población caucásica que en afroamericanos12. La edad media de presentación es de 63-75 años13. Existe una fuerte predisposición familiar, reportándose un riesgo de hasta 20 veces en desarrollar MW en pacientes con familiares de primer grado portadores de MW14. En México se tiene reportada una incidencia del 0.18% de todas las enfermedades hematológicas, siendo 11 veces menos frecuente que en la población caucásica15.
Aunque se considera una enfermedad incurable, la mediana de supervivencia libre de enfermedad ha mejorado, de seis años en la década de los 90 a ocho años a partir del año 200016.
Es importante diferenciarla de la gammapatía monoclonal de significado incierto (GMSI) tipo IgM, el MM, el linfoma del manto, la leucemia linfocítica crónica (LLC) y el linfoma de la zona marginal (LZM). Infecciones como hepatitis, por el VIH y varias enfermedades reumatológicas también elevan los niveles de IgM. La presencia de la mutación MYD88 L265P distingue la MW de otras enfermedades linfoproliferativas crónicas de células B17.
Patogénesis
Existen numerosas anormalidades y mutaciones genéticas, dentro de ellas las más comunes incluyen: del(6q) (50%), hipermutación somática en IGHV, t(9;14) (p13;q32) (50%) y trisomía 4 (20%). En la oncogénesis de la MW, las mutaciones somáticas en los genes MYD88 (>90%) y CXCR4 (30-35%) juegan un papel pivotal18.
El producto del gen de la respuesta primaria de diferenciación mieloide (MYD88) es una proteína adaptadora (conecta señales extracelulares a señales intracelulares) del receptor 4 toll-like y de los receptores de las IL 1 y 2. Puede ser activada directamente por estos receptores o por interacción de la proteína adaptadora del dominio TIR y la tirosinacinasa de Bruton, promoviendo la activación de la vía del factor nuclear kappa B. Existen numerosas variantes somáticas, siendo la MYD88 L265P (sustitución de leucina por prolina en la posición 265) la más frecuente, resultando en una ganancia de función19. Esta mutación es rara o ausente en la GMSI-IgM (10-60%), el LZM (7%), la LLC (3%) y el MM (0%)19,20.
El receptor de quimiocina tipo 4 (CXCR4) es un receptor de proteína G acoplada, la cual juega un papel importante en la liberación de citocinas y en la quimiotaxis. Las mutaciones en esta proteína resultan en una activación permanente por el factor derivado de estroma 1 alfa (SDF-1α). La supresión del CXCR4 resulta en la inhibición de la migración y adhesión de las células de la MW. Las mutaciones somáticas de este receptor se encuentran en un 27% de los pacientes con MW21; la mutación CXCR4 S338 ha mostrado respuestas inferiores al ibrutinib22.
Diagnóstico
El diagnóstico de la MW está basado en la confirmación histopatológica de infiltración a MO por un linfoma linfoplasmocítico (presencia de linfocitos pequeños con plasmocitos y linfoplasmocitos) y la detección de un componente monoclonal IgM en suero, el cual debe ser siempre confirmado por inmunofijación. Los cuerpos de Dutcher (vacuolas intranucleares que contienen IgM) se observan frecuentemente. La población monoclonal linfoplasmocítica en MO, mediante inmunofenotipo o inmunohistoquímica, debe expresar CD19, CD20, CD22 y CD79a en los linfocitos; CD38 para el componente plasmocítico; además de acumulación de células B con fenotipo CD22(+dim), CD25+, CD27+ e IgM+, que lo difiere de otros linfomas por su expresión negativa de CD5, CD10, CD11c o CD10323. El resto de la evaluación diagnóstica se resume en la tabla 3.
Tabla 3 Evaluación diagnóstica
| Evaluación diagnóstica |
|---|
| Recomendado |
| - Historia clínica y examen físico • Incluir historia familiar de MW y otras enfermedades linfoproliferativas |
| - Revisión por sistemas (síntomas B-14%),
organomegalias, síntomas de hiperviscosidad, neuropatía,
enfermedad de Raynaud, rash, edema periférico,
anormalidades dérmicas, disnea • Incluir fondoscopia en casos de IgM elevada y sospecha de síndrome de hiperviscosidad |
| - Estudios de laboratorio: • Biometría hemática completa • Panel metabólico completo • Panel de inmunoglobulinas (IgA, IgG, IgM) • b2-microglobulina • Serología viral (VIH, VHB, VHC) |
| - Aspirado de médula ósea y biopsia de hueso
• Inmunohistoquímica (indispensable) • Citometría de flujo (opcional, en caso de no contar con inmunohistoquímica) • Test de mutación del gen MYD88 - Biopsia de ganglio linfático u otro sitio linfoide involucrado |
| - TC de tórax, abdomen y pelvis (si clínicamente está indicado y en todos los pacientes considerados a inicio de tratamiento) |
| Opcionales |
| - Crioglobulinas - Viscosidad sérica - Screening de enfermedad de von Willebrand adquirida - Proteinuria de 24 horas - Cadenas ligeras séricas - NTproBNP, troponinas - Electromiografía (consultar con un neurólogo) |
MW: macroglobulinemia de Waldenström; Ig: inmunoglobulina; VIH: virus de la inmunodeficiencia humana; VHB: virus de la hepatitis B; VHC: virus de la hepatitis C; TC: tomografía computarizada; NTproBNP: fragmento aminoterminal del péptido natriurético tipo B.
Una vez realizadas las pruebas recomendadas para el diagnóstico de MW, se debe clasificar la etapa de la enfermedad con base en los criterios que se presentan en la tabla 4.
Tabla 4 Criterios diagnósticos para macroglobulinemia de Waldenström
| Criterios diagnósticos para macroglobulinemia de Waldenström |
|---|
| Gammapatía monoclonal IgM (independiente de la concentración) Infiltración de medula ósea por linfoplasmablastos Inmunofenotipo de MO: CD5, CD10, CD19+, CD20+, CD23 Descartar otras patologías de linfocitos maduros |
Ig: inmunoglobulina; MO: médula ósea.
Adaptada de Dispenzieri, 20172.
Manifestaciones clínicas
Las manifestaciones se pueden dividir en dos grandes grupos: involucramiento a órgano blanco y síntomas relacionados con la paraproteína IgM. El involucramiento a órgano blanco incluye: síntomas B (fiebre, pérdida ponderal, fatiga, diaforesis nocturna), linfadenopatía, hepatomegalia y/o esplenomegalia e infiltración a MO. Los síntomas relacionados con la paraproteína IgM incluyen: amiloidosis sistémica, crioglobulinemia, neuropatía periférica (20-25% de los casos) y síntomas relacionados a hiperviscosidad (cambios visuales, compromiso neurológico y cardiovascular, ocurriendo normalmente cuando los niveles están por encima de 30-40 g/dl)24.
Estratificación de riesgo y factores pronósticos
Un análisis multivariante realizado en 1,511 paciente registrados en la sobrevida libre de recaida (SLR) de enero del 2000 a diciembre del 2014 demostró que la edad, síntomas B, deshidrogenasa láctica (DHL) elevada y niveles de hemoglobina (Hb) < 11.5 g/dl fueron factores de mal pronóstico. El nivel sérico de IgM no tuvo significado pronóstico; asimismo, el sexo del individuo no presentó significancia pronóstica.
En el IPSS (International Prognostic Scoring System) descrito para la MW10, incluyen cinco variables que impactan negativamente en el pronóstico: anemia, trombocitopenia, niveles de β2-microglobulina, concentración sérica de IgM y la edad (Tabla 5).
Tabla 5 Estratificación de riesgo por IPSS MW 2009
| Estratificación de riesgo por IPSS MW 2009 | |||
|---|---|---|---|
| Grupo de riesgo | Bajo | Intermedio | Alto |
| Factores de riesgo* | 0-1 (excepto edad) | Edad o 2 | ≥ 3 |
| SG a 5 años | 87% | 68% | 36% |
*Factores de riesgo: edad ≥ 65 años, Hb ≤ 11.5 g/dl, plaquetas ≤ 100x109/l,
B2M > 3 mg/dl e IgM >70g/l.
IPPS: International Prognostic Scoring System; MW: macroglobulinemia de Waldenström; SG: supervivencia global; Hb: hemoglobina; B2M: microglobulina beta 2.
Adaptada de Dispenzieri, 20172
En el IPSS no se incluyó el nivel sérico de DHL y los bajos niveles de albúmina de importancia pronóstica adversa.
Tratamiento
Pacientes asintomáticos (watch-and-wait)
No existe un tratamiento estandarizado en enfermedad asintomática. En pacientes con MW asintomática se recomienda un seguimiento en frecuencia de 3-4 meses, en caso de enfermedad estable se puede extender hasta cada seis meses. Dentro de la evaluación se deben considerar los siguientes puntos: examen físico detallado, incluida la fondoscopia (preferentemente valorada por un oftalmólogo) para valorar congestión de la vena retiniana, hemorragias o exudados y papiledema.
Criterios de inicio de tratamiento
Deberá iniciarse tratamiento en aquellos pacientes con niveles de IgM > 60 g/l, síntomas constitucionales, hepatoesplenomegalia sintomáticos o linfadenopatías, ya sea sintomático o bulky (> 5 cm), síndrome de hiperviscosidad, neuropatía severa, amiloidosis sistémica o crioglobulinemia sintomática, Hb ≤10 mg/dl y plaquetas < 100x109,25.
Pacientes sintomáticos
En la MW no existe un estándar de tratamiento, la falta de ensayos aleatorizados entre los regímenes hace que sea difícil proporcionar recomendaciones con nivel de evidencia A. Las opciones de tratamiento se basan en gran medida en la experiencia a largo plazo y la opinión de expertos. La inmunoquimioterapia multiagente debe ser preferida sobre el rituximab en monoterapia, ya que ha mostrado tasas de respuesta parcial del 80%, en comparación con el rituximab, donde las tasas de respuesta son < 50%.
La hiperviscosidad es una manifestación infrecuente de la MW, la presentación más común es el sangrado nasal o gingival. El nivel de viscosidad siempre debe medirse en pacientes con síndrome de hiperviscosidad sospechoso, siendo la terapia de elección el intercambio plasmático. Un solo intercambio de plasma puede normalizar la viscosidad y permitir que la quimioterapia reduzca con éxito la masa tumoral. Uno de los inconvenientes de la monoterapia con rituximab es el lento tiempo de respuesta y el incremento transitorio de IgM o flare, el cual ocurre dentro del 1.er y 4.o mes de tratamiento en un 10-15% de los casos, con riesgo de exacerbar la neuropatía y la sintomatología asociada a hiperviscosidad. Además, se requiere precaución en diferenciar un flare de IgM de una falta de respuesta.26
Los regímenes de tratamiento combinado con rituximab han demostrado una reducción rápida de los niveles de IgM, siendo utilizados en pacientes con hepatoesplenomegalia o infiltración significativa a MO y que requieren una rápida citorreducción.
La combinación de rituximab con ciclofosfamida y dexametasona mostró su efectividad en un estudio prospectivo de 72 pacientes näive, dando como resultado una tasa de respuesta global (TRG) del 83%; la supervivencia libre de progresión (SLP) y la SG a dos años del 67 y el 81%, respectivamente.27 En la actualización de este estudio, la media de tiempo para recaída fue de 35 meses; en relación con grupos de riesgo, la SG a ocho años fue de 100, 55 y 27% para los grupos de riesgo bajo, intermedio y alto, respectivamente.28
En un estudio aleatorizado de 64 pacientes se comparó el uso de CHOP (ciclofosfamida + doxorubicina + vincristina + prednisolona) vs. rituximab-CHOP (R-CHOP), demostrando este último régimen una mayor respuesta global (RG) (91 vs. 60%) y SLP a dos años (78 vs. 47%). La fludarabina ha demostrado respuesta significativa en combinación con rituximab (FR), así como en combinación con este y ciclofosfamida (FCR). El esquema FR demostró una TRG y una SLP en dos años del 96.3 y el 67%, respectivamente29,30. El esquema FCR consiguió, en un estudio retrospectivo, una TRG y una SG a tres años del 76 y 96%, respectivamente31. Los esquemas asociados con fludarabina se recomiendan en pacientes en recaía o refractarios, debido a las citopenias prolongadas y al riesgo de segundas neoplasias (10-15%). El uso combinado de rituximab con bendamustina (BR) demostró mejores resultados frente a R-CHOP en un ensayo clínico fase III, alcanzando mayor SLP (69.5 vs. 28.1 meses; p < 0.001) con un mejor perfil de seguridad.32
El uso de IP, al igual que en pacientes con MM, ha demostrado respuesta significativa en pacientes con MW. En un ensayo fase II33 con 26 pacientes, el uso combinado de bortezomib con rituximab alcanzó una Respuesta Menor o mejor en el 88%, con una reducción de IgM en el 100% de los pacientes; la mediana de SLP no se alcanzó, con una estimación al año de SLP del 75%; la SG no se alcanzó, con un estimado a un año del 96%. El esquema con rituximab y dexametasona (BRD) ha demostrado su eficacia en dos estudios, alcanzando una TRG del 90-96%, una SLP del 40-80% y una SG del 80-100%. Sin embargo, la toxicidad más frecuente fue la neuropatía, requiriendo la suspensión del bortezomib en el 8-61% de los pacientes34,35.
En un estudio fase III en pacientes previamente tratados, el uso de carfilzomib alcanzó una TRG del 87.1%, presentándose neuropatía en menor frecuencia (3.2%) y sin ser ≥ grado 3. El carfilzomib, a partir de 20 mg/m2 y a escala de 36 mg/m2, se recibió los días 1, 2, 8, 9 de cada ciclo; el rituximab y la dexametasona en los días 2 y 9 de cada ciclo, y un tratamiento de mantenimiento cada ocho semanas por ocho ciclos. Como respuesta general, la tasa en 31 pacientes fue del 87%; el 36% logró una muy buena respuesta parcial (VGPR) con un tiempo medio de respuesta de 2.1 meses y sin neuropatía periférica > grado 1. Esta combinación también se ha utilizado para el tratamiento de la enfermedad en recaída36.
Recaída o enfermedad refractaria
El uso de rituximab ha demostrado eficacia en pacientes con recaída o refractariedad. En un ensayo fase II se incluyeron 72 pacientes (34 previamente tratados), obteniéndose una respuesta similar entre los pacientes tratados y los no tratados (51.4 y 52.9%, respectivamente). La combinación de rituximab con fludarabina o brentuximab ha demostrado mejores respuestas, con una TRG del 83.3 y el 93.8%, respectivamente. En un estudio retrospectivo, el esquema FCR demostró una TRG y una SG a tres años del 77.2 y 89%, respectivamente.33
Los IP han demostrado su efectividad en diferentes estudios, inicialmente estudios con bortezomib en monoterapia. En un pequeño estudio de 10 pacientes36 se obtuvo una respuesta global y mayor del 80 y el 60%, respectivamente. En otros dos estudios de mayor población el bortezomib en monoterapia alcanzó respuestas globales y respuestas mayores del 78-85 y 44-48%, respectivamente. En ambos estudios el tiempo para alcanzar respuesta fue rápido (1.4 y 1.5 meses) y la duración de la respuesta variable, con una media de progresión de 6.6 meses en un estudio y una SLP de 16 meses en otro37. En un estudio fase II más reciente se evaluó el bortezomib en monoterapia y en aquellos casos con falta de respuesta se agregó manejo con dexametasona (20 mg/día por 2 días); la TRG fue del 43.2%, con una SG a dos años del 84% y una mediana de SLP de 15.3 meses, sin diferencias entre los grupos a los que se les agregó o no dexametasona.
El uso combinado de bortezomib con otros esquemas de tratamiento ha mejorado la profundidad de la respuesta y la SLP. En un estudio prospectivo, el esquema BDR ((bortezomib + dexametasona + rituximab) demostró efectividad, con una TRG y respuesta mayor del 96 y el 83%, respectivamente; el tiempo para alcanzar respuesta fue rápido (1.4 meses). En el seguimiento de este mismo estudio, la RG y la respuesta mayor fue del 96 y el 91%, respectivamente, con una media de progresión de 52 meses. Sin embargo, la principal toxicidad fue la neuropatía relacionada a uso de bortezomib, requiriendo su suspensión hasta en el 60% de los pacientes. En cuanto a la manera de mejorar la neurotoxicidad, Ghobrial, et al., en dos estudios similares administraron esquemas a diferentes dosis, obteniendo una TRG y una respuesta mayor del 81-88 y 51-65%, respectivamente; la neuropatía periférica se presentó en el 41% grado 1-2 y solo en el 5% grado ≥ 3.37
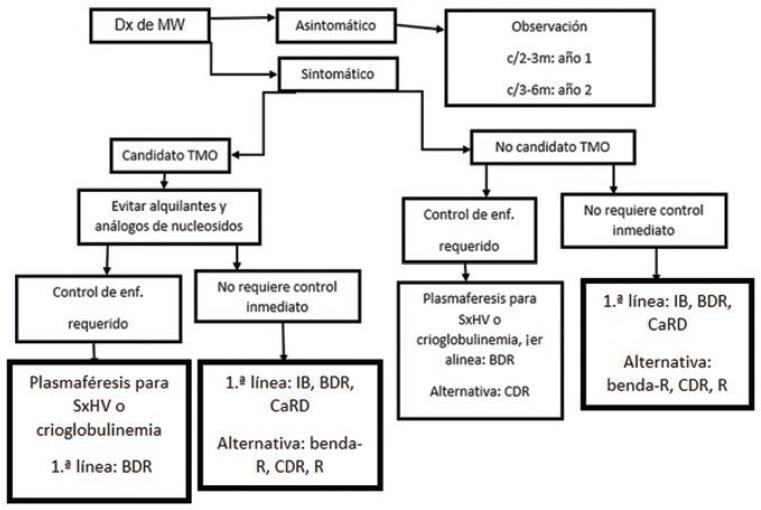
Figura 1 Adaptado de Gertz MA.2018.26Dx: diagnóstico; MW: macroglobulinemia de Waldenström; TMO: trasplante de médula ósea; SxHV: Síndrome de Hiperviscosidad; benda-R: bendamustina + rituximab; BDR: bortezomib + dexametasona + rituximab; CaRD: Carfilzomob-Lenalidomida-Dexametasona; CDR: Carfilzomib-Daratumumab-Lenalidomida; R: rituximab.

Figura 2 Adaptado de Gertz MA.2018.26IgG: inmunoglobina G; Hb: hemoglobina; tto: tratamiento; CPH: Células Progenitoras Hematopoyéticas; Cfa: Ciclofosfamida.
El uso de carfilzomib a un bajo riesgo de neuropatía periférica, pero con un mayor riesgo de cardiotoxicidad se ha evaluado en una serie de casos (p = 7) en combinación con dexametasona en pacientes con recaída, obteniéndose una respuesta global y una respuesta mayor del 100 y el 86%, respectivamente.
El inhibidor de la diana de rapamicina en células de mamífero everolimús se ha utilizado para tratar la MW en un ensayo con 60 pacientes, quienes fueron tratados con recaída o refractarios, con una tasa de respuesta global del 50% y una tasa de beneficio clínico de 73%. La mediana del tiempo de respuesta para pacientes que lograron una respuesta parcial (PR) fue de dos meses. La mediana de SLP fue del 21%. Desafortunadamente, se observaron toxicidades grado 3 o superior en el 67% de los pacientes. El everolimús también se ha combinado con bortezomib y rituximab en un ensayo de fase I-II donde se administraron seis ciclos, seguidos de mantenimiento con everolimús; el 57% de los pacientes tenían bortezomib previo y el 98% rituximab previo, pero la tasa de SG a un año fue del 89%, la tasa de PR fue del 53% y la mediana de SLP fue de 21 meses26.
Mantenimiento
Castillo, et al. incluyeron 182 pacientes con tres regímenes de tratamiento: benda-R (bendamustina + rituximab), BDR, Ciclo (ciclofosfamida), dexa-R (dexametasona + rituximab), seguidos o no de mantenimiento con rituximab para pacientes sintomáticos. Los pacientes que lograron mayor supervivencia libre de enfermedad fueron los tratados bajo régimen de BDR, seguidos de mantenimiento con rituximab con un valor de p < 0.001.38
Tras la aplicación del tratamiento deberá evaluarse el grado de respuesta logrado con este, con base en los criterios descritos en la tabla 6.
Tabla 6 Criterios de respuesta del International workshop on Waldenström macroglobulinemia
| Criterios de respuesta del International workshop on Waldenström macroglobulinemia | ||
|---|---|---|
| Respuesta | Abreviación | Criterios |
| Respuesta completa | RC | IgM normal, desaparición de proteína anormal por inmunofijación, resolución de adenomegalias y visceromegalias, asintomático |
| Muy buena respuesta parcial | MBRP | Disminución > 90% de IgM sérica o de tamaño de adenomegalias y visceromegalias. Sin nuevos síntomas |
| Respuesta parcial | RP | Reducción > 50% de IgM sérica o de tamaño de adenomegalias y visceromegalias. Sin nuevos síntomas |
| Respuesta menor | RM | Reducción ≥ 25% pero < 50% de IgM sérica. Sin síntomas nuevos |
| Enfermedad estable | EE | Reducción < 25% y aumento < 25% de IgM o de adenomegalias |
| Enfermedad en progresión | EP | Aumento ≥ 25% de IgM o sintomatología o complicaciones atribuibles a MW |
Ig: inmunoglobulina; MW: macroglobulinemia de Waldenström.
Adaptada de Blood, et al., 201531.
Manifestaciones clínicas
Las manifestaciones clínicas del mieloma múltiple (MM) se pueden dividir en dos: a) aquellas secundarias a la actividad de la enfermedad y que son parte del acrónimo CRAB (hipercalcemia, insuficiencia renal, anemia y lesiones líticas); y b) las que son consecuencia de la alta carga tumoral, como las relacionadas a hiperviscosidad o plasmocitomas.
La gran mayoría de los pacientes con el diagnóstico final de MM llegan al hematólogo después de un largo camino a través de diversas especialidades y posterior al estudio de complicaciones como fracturas patológicas, sintomatología relacionada a insuficiencia renal y anemia. La temprana referencia de estos pacientes a nuestra consulta depende de la experiencia y conocimiento del médico de primer contacto, quien debe tener en mente las manifestaciones clínicas de la enfermedad para poder sospecharla.
Los datos más frecuentes son: anemia (73%), dolor óseo (52%), fatiga (32%), elevación de creatinina (48%), hipercalcemia (28%) y pérdida de peso (24%); la fiebre se presentó en solo el 0.7% de los casos1.
Criterios diagnósticos
Los criterios diagnósticos para gammapatía monoclonal de significado incierto (MGUS), MM asintomático y otras gammapatías se abordarán de manera exclusiva en el apartado que corresponde; en este apartado nos enfocaremos solamente en los criterios de MM.
Las gammapatías son enfermedades de compleja biología que requieren de una serie de estudios para integrar los diversos diagnósticos. El abordaje diagnóstico inicial del mieloma debe hacerse con base en la sospecha clínica y apoyado en los siguientes estudios de laboratorio y gabinete:
‒ Componente clínico: síndrome anémico, dolor óseo, letargo, cefalea u otro dato de síndrome de hiperviscosidad.
-
‒ Componente bioquímico:
Biometría hemática completa, glucosa, creatinina, nitrógeno ureico en sangre, globulinas, albúmina, proteínas totales, deshidrogenasa láctica, alanina aminotransferasa, aspartato aminotransferasa, ácido úrico, β2-microglobulina, calcio sérico y depuración de creatinina en orina de 24 h.
Determinación de niveles séricos de inmunoglobulinas (Ig).
Detección y evaluación del componente monoclonal con electroforesis en suero y/u orina.
Inmunofijación para determinación de restricción de cadenas ligeras.
‒ Componente morfológico: aspirado de medula ósea y biopsia.
‒ Componente de imagen: serie ósea metastásica, tomografía computarizada y resonancia magnética.
Habiendo realizado todos estos estudios estaremos en posibilidad de integrar alguno de los diagnósticos de gammapatías, específicamente para MM, del International Myeloma Working Group en su revisión del 2014 (Tabla 1).
Tabla 1 Criterios international Myeloma Working Group
| Se deben cumplir ambos criterios |
|---|
| - Células plasmáticas clonales > 10% o biopsia de hueso afectada o plasmocitoma extramedular |
| - Uno o más de los siguientes eventos definitorios:
• Evidencia de daño a órgano blanco que puede ser atribuido al desorden proliferativo de las células plasmáticas, específicamente: • Hipercalcemia: calcio sérico > 0.25 mmol/l (> 1 mg/dl) mayor al límite superior normal o > 2.75 mmol/l (> 11 mg/dl) • Insuficiencia renal: depuración de creatinina < 40 ml/min o creatinina sérica > 177 µmol/l (> 2 mg/dl) • Anemia: disminución de hemoglobina > 2 g/dl por debajo del límite normal o nivel de hemoglobina < 10 g/dl • Lesiones óseas: una o más lesiones osteolíticas por radiografía, TC o TC-PET • Células plasmáticas clónales en medula ósea > 60% • Afección en la relación de FLC > 100 • Más de 1 lesión lítica en RM (mínimo de 5 mm) |
Nota: de acuerdo con el componente monoclonal encontrado, el orden de frecuencia es: IgG 52%, IgA 21%, cadenas ligeras 16%, IgD 2%, biclonal 2%, IgM 0.5% y no secretor 6.5%.
TC: tomografía computarizada; TC-PET: tomografía computarizada-tomografía por emisión de positrones; RM: resonancia magnética; FLC: Free Light Chains.
Adaptada de Kyle, et al., 20031.
Estadificación y pronóstico
El primer paso para estadificar una enfermedad es diferenciar entre factores pronóstico y factores predictivos. Los primeros se refieren a factores que nos aportan información acerca de cómo evolucionará la enfermedad, mientras que los segundos nos orientan acerca de qué tan factible es que la enfermedad responda a una intervención terapéutica dada2. Habiendo aclarado esto, se debe diferenciar entre los factores clínicos y biológicos.
Tradicionalmente el MM había sido estadificado con base en la clasificación de Durie-Salmon, la cual toma en consideración factores clínicos y bioquímicos, dividendo a la enfermedad en tres categorías (riesgo bajo, intermedio y alto) según la presencia de lesiones líticas, el componente monoclonal, la hemoglobina y el calcio sérico, además, subdividiendo a los pacientes en dos grupos de acuerdo con su función renal. Este sistema ha caído en desuso debido a su poca utilidad y aplicabilidad con las nuevas técnicas de imagen3.
Actualmente el sistema más utilizado es el International Staging System (ISS), el cual requiere de dos marcadores bioquímicos relativamente accesibles y categoriza a la enfermedad en tres categorías de acuerdo con los niveles de β2-microglobulina y albúmina4 (Tabla 2).
Tabla 2 Índice pronóstico internacional
| Índice pronóstico internacional | ||
|---|---|---|
| Estadio | Criterio | Supervivencia global |
| I II III | β2-microglobulina < 3.5 y albumina sérica > 3.5 g/dl
Ninguno de I ni III β2-microglobulina > 5.5 |
62 meses 44 meses 29 meses |
Adaptada de Moreau, et al., 20174.
La aplicación de factores pronósticos bioquímicos, moleculares y genéticos ha cambiado radicalmente la forma en como entendemos al mieloma y, por lo tanto, también su abordaje desde diversos puntos de vista.
El ISS revisado es un sistema que toma en consideración los factores tradicionales del ISS y adiciona factores citogenéticos5.
Riesgo citogenético
Con el advenimiento de nuevas tecnologías, las alteraciones citogenéticas tomaron fuerza en la determinación del pronóstico de esta enfermedad. Estas técnicas ya habían demostrado su utilidad en otras enfermedades hematológicas, por lo cual solo era cuestión de tiempo para que se demostrara su aplicación al mieloma. Inicialmente, el cariotipo fue la herramienta utilizada para este fin, para posteriormente dar paso a las técnicas de hibridación in situ (FISH). El panel de alteraciones debe contener por lo menos las que se enlistan en la tabla 3.
Tabla 3 Anormalidades citogenéticas y curso clínico
| Anormalidad citogenética | Estadio clínico de detección |
|---|---|
| Mieloma múltiple | |
| Trisomías | Buen pronóstico, riesgo estándar, SG 7-10 años |
| t(11:14)(q13;q32) | Buen pronóstico, riesgo estándar, SG 7-10 años |
| t(6:14)(p21;q32) | Buen pronóstico, riesgo estándar, SG 7-10 años |
| t(4:14)(p16;q32) | Riesgo intermedio, SG 5 años |
| t(14:16)(q32;q23) | Riesgo alto, SG 3 años, relacionado a alto índice de cadenas ligeras libres e insuficiencia renal inicial |
| t(14:20)(q32;q11) | Riesgo alto, SG 3 años |
| Ganancia (1q21) | Riesgo intermedio, SG 5 años |
| Del(17p) | Riesgo alto, SG 3 años |
| Normal | Buen pronóstico, SG 7-10 años |
Adaptada de Rajkumar, 20165.
Las traslocaciones relacionadas con el cromosoma 14 [t (4:14), t (11:14), t (6:14), t(14:16) y t(14:20)] resultan en productos de fusión relacionados a la cadena pesada de la Ig, lo que estimula a una serie de oncogenes que aumentan la capacidad proliferativa de la clona alterada. La deleción del brazo corto del cromosoma 17 (Del17p) está relacionada a la pérdida del gen supresor TP533.
Como se mencionó anteriormente, la combinación de factores clínicos, bioquímicos y citogenéticos mejora la capacidad de predicción y pronóstico global de estos factores, surgiendo así el esquema ISS revisado (Tabla 4), el cual es independiente de la edad del paciente y la terapia recibida, dando mejor información pronóstica que el ISS. La Mayo Clinic ha desarrollado también un estadiaje citogenético que nos orienta acerca del pronóstico de los pacientes de reciente diagnóstico (Tabla 5).
Tabla 4 International Staging System (ISS) revisado
| Estadio | Supervivencia global a 5 años | Supervivencia libre de progresión a 5 años |
|---|---|---|
| ISS-R I | 82% | 55% |
| ISS-R II | 62% | 36% |
| ISS-R III | 40% | 24% |
| International Staging System revisado | ||
|
Estadio I
Todos los siguientes criterios: - Albúmina sérica ≥ 3.5 mg/dl - β2-microglobulina sérica < 3.5 mg/dl - Sin alteraciones citogenéticas de alto riesgo - LDH sérica normal | ||
|
Estadio II
- No cumple criterios para estadio I o III | ||
|
Estadio III
Ambos criterios: - β2-microglobulina sérica > 5.5 mg/dl - Alteraciones citogenéticas de alto riesgo [t (4:14) o Del17p] o LDH elevada | ||
LDH: lactato deshidrogenasa.
Adaptada de Rajkumar, 20165.
Tabla 5 Estratificación de riesgo citogenético de la Mayo Clinic
| Estratificación de riesgo citogenético de la Mayo Clinic | |
|---|---|
| Grupo de riesgo | % de pacientes de novo con la alteración |
| Riesgo estándar | 75% |
| Trisomías | |
| t(11;14) | |
| t(6:14) | |
| Riesgo intermedio | 10% |
| t(4:14) | |
| Ganancia (1q) | |
| Riesgo alto | 15% |
| t(14:16) | |
| t(14;20) | |
| Del(17p) | |
Adaptada de Dimitros, et al., 20183.
Módulo 4: Tratamiento
El tratamiento del mieloma múltiple ha cambiado radicalmente durante los últimos 10 años. La inclusión de nuevos agentes y combinaciones ha logrado que los médicos consigan diseñar los tratamientos de acuerdo con las características y necesidades de cada paciente, sin embargo, la gran cantidad de información generada requiere de un análisis preciso y objetivo para tomar la mejor decisión en cada caso. Son diversas las guías de tratamiento internacionales que se han desarrollado y actualizado en esta última década, la información que en ellas se plasma es de gran valor, pues orienta objetivamente al clínico en su diario que hacer frente al paciente con mieloma múltiple.
De manera objetiva, hemos dividido las distintas opciones terapéuticas agrupando a los pacientes con base en: edad, estado físico, riesgo citogenético y líneas de tratamiento recibidas, de esta forma es más sencilla la toma de decisiones.
Tratamiento de primera línea para pacientes menores de 65 años, buen desempeño físico y/o candidatos a TCPH, riesgo estándar
Este grupo de pacientes representa una gran proporción en centros médicos donde se realiza el protocolo de abordaje diagnóstico para esta enfermedad, mientras que en los centros de referencia se encuentra un bajo porcentaje. El tratamiento de primera línea de estos pacientes debe tener como objetivo lograr la mejor respuesta posible previa al trasplante de células progenitoras hematopoyéticas (TCPH), esto mediante la combinación de agentes con distintos mecanismos de acción que aseguren la cobertura de diversos puntos patológicos de la enfermedad para, con ello, propiciar una respuesta profunda.
Son varios los estudios que han demostrado que la combinación de tres fármacos con distintos mecanismos de acción logra este objetivo. En un metaanálisis publicado por un grupo brasileño, en el cual se estratificaron los esquemas de tratamiento disponibles en la literatura científica desde 2006 hasta 2016 agrupándolos en aquellos basados en antracíclicos, inhibidores del proteosoma (IP) e inmunomoduladores (IMiD), se encontró que los que presentaron mejores tasas de respuesta eran los que incluían IMiD e IP, y en último lugar los que están basados en antracíclicos. Los perfiles de seguridad de acuerdo con la combinación elegida pueden guiar cada tratamiento de acuerdo con las comorbilidades de cada paciente1.
Las guías europeas y estadounidenses reproducen la indicación de combinar tres fármacos para este grupo específico, incluyendo esquemas con combinaciones de IP, IMiD y quimioterapia, haciendo consideraciones especiales para grupos específicos que se discutirán en otros apartados. Con base en estas recomendaciones determinamos los siguientes lineamientos:
‒ En pacientes menores de 65 años, con buen desempeño físico y candidatos a TCPH se deberá iniciar tratamiento con esquemas de tres fármacos que incluyan al menos un IP/IMiD, las posibles combinaciones se resumen en la figura 1 y figura 2.
‒ Posterior a cuatro ciclos se debe determinar respuesta de la enfermedad, siguiendo los criterios que se enumeran en el apartado Criterios de respuesta.
‒ Seguir los lineamientos expuestos en el apartado Trasplante autólogo de células progenitoras hematopoyéticas para solicitar su inclusión a lista de espera del programa de trasplante.
‒ La enfermedad mínima residual tiene un valor agregado para el clínico en la toma de decisiones para el número de ciclos que el paciente debe recibir.

Figura 1 Esquema de tratamiento ideal para los pacientes con mieloma múltiple, de acuerdo con la elegibilidad para trasplante (adaptada de Kumar, et al., 20122).TCPH: trasplante de células progenitoras hematopoyéticas.

Figura 2 Algoritmo de tratamiento en candidatos a trasplante.TCPH: trasplante de células progenitoras hematopoyéticas; VCD: Bortezomib-Ciclofosfamida-Dexametasona; VTD: Bortezomib-Talidomida-Dexametasona; VRD: Bortezomib-Lenalidomida-Dexametasona; KRD: Carfilzomib-Lenalidomida-Dexametasona.Adaptada de Dingli, et al., 2017 4.
El seguimiento de estos pacientes se hará de acuerdo con los lineamientos expuestos en el apartado Criterios de respuesta y seguimiento.
Actualmente existen ya estudios que han analizado la eficacia de esquemas con talidomida vs. aquellos con ciclofosfamida, demostrando mayores tasas de respuesta completa y respuesta completa estricta en los esquemas que incluyen talidomida. Sin embargo, la alta incidencia de eventos adversos neurológicos y trombóticos al compararlo con ciclofosfamida debe tomarse en consideración previamente a su indicación2. Se deberán considerar también esquemas especiales para este grupo de pacientes, como son aquellos con alguna contraindicación para recibir IP, en los cuales esquemas como lenalidomida-dexametasona pueden utilizarse, con buenas tasas de respuesta en comparación con esquemas tradicionalmente utilizados, como melfalán o talidomida. Los esquemas utilizados por consenso internacional para aquellos pacientes que requieren quimioterapia intensiva (leucemia de células plasmáticas o enfermedad extramedular) recomiendan como una opción el VTD-PACE (bortezomib, dexametasona, talidomida, cisplatino, doxorubicina, ciclofosfamida y etopósido), sin embargo, y debido a la alta toxicidad relacionada con el tratamiento de este esquema, se debe discutir y valorar de manera integral al paciente antes de decidir por esta rama terapéutica3.
Pacientes mayores de 65 años o con mal estado físico, no candidatos a TCPH
Las escalas de fragilidad han permitido unificar criterios para la elección de los pacientes no candidatos a trasplante. En este grupo de pacientes se debe considerar el uso de los nuevos agentes y combinaciones, ya que han demostrado aumento en la supervivencia, con respuestas más profundas y mejor calidad de vida.
Es por ello que en pacientes no candidatos a TCPH, ya sea por edad o por mal estado físico, consideramos lo siguiente:
‒ Utilizar combinaciones que incluyan un IP y un IMiD por al menos ocho ciclos del esquema seleccionado. Las posibles combinaciones se resumen en la (Figura 3).
‒ Realizar valoración de la enfermedad siguiendo los lineamientos del apartado Criterios de respuesta.
‒ Posteriormente a la conclusión de los ciclos mínimos de tratamiento de acuerdo con el esquema elegido, continuar con mantenimiento.

Figura 3 Algoritmo de tratamiento para no candidatos a trasplante.TCPH: trasplante de células progenitoras hematopoyéticas; VCD: Bortezomib-Ciclofosfamida-Dexametasona; VTD: Bortezomib-Talidomida-dexametasona; VRD: Bortezomib-Lenalidomida-Dexametasona; MPT: Melfalán-Prednisona-Talidomida; DMVD: Daratumumab-Melfalán-Bortezomib-Dexametasona LD: Lenalidomida-dexametasona; BD: Bortezomib-Dexametasona ; KRD: Carfilzomib-Lenalidomida-dexametasona.Adaptada de Dingli, et al., 2017 4.
De igual manera que en aquellos candidatos a TCPH, el uso de esquemas alternativos puede considerarse en casos especiales, como intolerancia a IMiD o a IP, neuropatía previa o contraindicación por parte de alguno de los servicios de apoyo interconsultados (p. ej., cardiopatía, nefropatía grave o demencia). En estos casos se considerará el uso de esquemas con dos fármacos, de acuerdo con el perfil de cada paciente, asi como la adaptación en la forma de administrar el medicamento a utilizar de acuerdo a sus distintos perfiles de seguridad y toxicidad (Tablas 1 y 2).
Tabla 1 Vías de administración, dosis, eventos adversos y consideraciones especiales de los inhibidores del proteasoma
| Bortezomib | Carfilzomib | Ixazomib | |
|---|---|---|---|
| Vías de administración | SC | IV | VO |
| Dosis y esquemas | 1.3 mg/m2 en los días 1, 4, 8 y 11 Ciclos de 28 días | 20/27 mg/m2 en los días 1, 2, 8, 9, 15 y 16 o 20/70 mg/m2 semanal por 3 semanas Ciclos de 28 días | 4 mg en los días 1, 8 y 15 Ciclos de 28 días |
| Eventos adversos que vigilar | - Neuropatía periférica - Hipotensión - Toxicidad cardiaca - Toxicidad pulmonar - Toxicidad gastrointestinal -Trombocitopenia - Neutropenia |
- Insuficiencia cardiaca - Insuficiencia renal - Toxicidad pulmonar y disnea - Hipertensión - Trombosis venosa - Hemorragia - Trombocitopenia - Toxicidad hepática |
- Trombocitopenia - Toxicidad gastrointestinal - Neuropatía periférica - Rash - Hepatotoxicidad |
| Consideraciones de manejo | Monitorizar cuenta plaquetaria, seguro en Insuficiencia renal | Vigilar función cardiaca y renal | Reducir dosis en insuficiencia renal |
SC: subcutánea; IV: intravenosa; VO: vía oral.
Tabla 2 Vías de administración, dosis, eventos adversos y consideraciones especiales de los fármacos inmunomoduladores
| Lenalidomida | Pomalidomida | Talidomida | |
|---|---|---|---|
| Vía de administración | VO | VO | VO |
| Dosis y esquemas | 25 mg/día 21 días con semana de descanso (inducción) 10 mg/día para mantenimiento | 4 mg/día 21 días con semana de descanso | 100 mg diarios |
| Eventos adversos que vigilar | - Tromboembolismo venoso - Trombocitopenia - Neutropenia - Fatiga - Rash - Pobre movilización de CPH - Segundas neoplasias |
- Tromboembolismo venoso - Neutropenia - Fatiga - Hepatotoxicidad - Rash |
- Tromboembolismo venoso - Constipación - Neuropatía periférica - Hipotensión ortostática - Rash - Bradicardia - Somnolencia |
| Consideraciones de manejo | Vigilar función renal (ajuste de la dosis en tratamiento con lenalidomida) Profilaxis para trombosis según factores de riesgo individuales o factores relacionados con el mieloma ASA (81-325 mg/día) o HBPM (enoxaparina 40 mg/día) o warfarina (INR: 2-3) | ||
VO: vía oral; ASA: ácido acetilsalicílico; HBPM: heparina de bajo peso molecular; INR: international normalized ratio; CPH: células progenitoras hematopoyéticas.
Tratamiento segunda línea y progresión en candidatos y no candidatos a TCPH
El manejo de las recaídas en estos pacientes implica un análisis más profundo de la enfermedad, el paciente y su respuesta al tratamiento o tratamientos previos. Es en este apartado donde toman mayor importancia factores relacionados con mal pronóstico que determinarán, en cierta medida, qué terapia debemos elegir para nuestro paciente. Cabe señalar que un punto básico del tratamiento de recaída es lograr la mejor respuesta posible, independientemente de ser candidato o no a trasplante.
La elección del régimen de tratamiento a la recaída es complicada, ya que se deben valorar muchos factores como: el tiempo a la recaída, la respuesta y profundidad alcanzada con la terapia previa, la agresividad de la recaída y el desempeño físico. Por tal motivo, es importante la selección del esquema de rescate en pacientes con recaída, ya que impactará en el periodo libre de progresión, debido a que la duración de las respuestas en las remisiones subsecuentes será menor a la primera respuesta alcanzada4.
La progresión se debe tratar inmediatamente cuando sea identificada, en la tabla 3 se enumeran los criterios que obligan a tratar a un paciente.
Tabla 3 Criterios del International Myeloma Working Group (IMWG) para progresión del mieloma múltiple (MM)
| Criterios del IMWG para progresión de MM | ||
|---|---|---|
| Recaída no agresiva | Recaída agresiva | |
| Bioquímica | Clínica | Anormalidades citogenéticas adversas: t(4:14), del (17p) |
| Incremento proteína M | Síntomas leves y elevación leve de proteína M | Elevación de b2-microglobulina o disminución de albúmina, enfermedad extramedular, aumento de LDH |
| Asintomático | Compromiso orgánico importante | Corta duración de la respuesta lograda |
| Presentación clínica agresiva | ||
| CP circulantes | ||
| Cambio en clona o isotipo de inmunoglobulina o cadena ligera | ||
LDH: lactato deshidrogenasa; CP: células plasmáticas.
Adaptada de Sonneveld, et al., 20175.
Es adecuado considerar los siguientes puntos para iniciar el tratamiento:
‒ Características del paciente en recaída. Se debe tomar en cuenta el desempeño físico del paciente, así como su elegibilidad para TCPH. En aquellos pacientes añosos y frágiles los esquemas tendrán como objetivo la mejor respuesta posible, pero también la menor toxicidad posible.5 Las combinaciones utilizables para este grupo de pacientes pueden ser aquellas que incluyen IP, IMiD y agente alquilante, como el melfalán. El International Myeloma Working Group sugiere que se utilicen esquemas cuya toxicidad conocida, sea lo menos impactante posible. En pacientes en buenas condiciones debemos tomar en consideración los deseos y necesidades en otros ámbitos de la vida, es decir, si el paciente es capaz de acudir a recibir su terapia intravenosa establecida o prefiere o necesita un esquema que sea vía oral, siempre teniendo en mente el logro de la respuesta más profunda.5
‒ Características de la enfermedad. Toman relevancia los factores de riesgo conocidos para recaída y mala respuesta al tratamiento. Alteraciones como la del17p pudieron haber estado ausentes al momento del diagnóstico e identificarse en la valoración de la progresión, de igual manera existen pacientes con riesgo estándar al diagnóstico que recaen con características de alto riesgo.6 Fue hasta hace poco que se les concedió valor a estos factores para la toma de decisiones, de esta manera actualmente se prefiere el uso de esquemas más agresivos, para aquellos que lo toleren, cuando se tiene alguno de estos datos presentes. La combinación de tres fármacos, incluyendo IP de segunda generación e IMiD de segunda generación se prefiere sobre el retratamiento con el esquema previo debido a lo observado en diversos estudios fase II, en los cuales se administraron esquemas basados en carfilzomib, lenalidomida u otros agentes novedosos a pacientes con estas características.
‒ Características de la respuesta previa. Se puede considerar retratamiento con el esquema anterior siempre y cuando la respuesta previa haya tenido una respuesta parcial y una duración de por lo menos 18 meses o intervalo libre de tratamiento de 6 a 9 meses, tomando en consideración efectos adversos y toxicidades secundarias al tratamiento. En caso de que la progresión haya ocurrido durante el tratamiento o con una duración menor a los 6 meses se debe considerar como paciente de alto riesgo y tratarse como dicho grupo . En aquellos casos en los que no se logre una respuesta profunda durante el retratamiento se puede escalar uno de los agentes utilizados antes de decidir cambio completo de combinación.6
‒ Elegibilidad para trasplante. Aquellos pacientes que no han sido sometidos a TCPH deberán ser tratados en busca de una profunda respuesta para ser llevados a dicho procedimiento, basado en combinación de tres fármacos. En general se considera un segundo esquema de tratamiento y segundo trasplante en aquellos sometidos previamente a uno y que hayan tenido una supervivencia libre de progresión de 18 a 24 meses o más. En caso de que estos pacientes no hayan recibido esquema de consolidación posterior al primer trasplante, se deberá considerar posterior al segundo trasplante.6
Tercera línea y subsecuente
Conforme aumentan las líneas de tratamiento, las posibilidades de respuesta disminuyen en un promedio del 5% por cada esquema.
Los esquemas para tercera línea o posteriores deben tomar en consideración las mismas características que se tomaron en la recaída anterior, teniendo en cuenta el desempeño físico del paciente a dos o tres líneas de tratamiento, ya que pudo haber tenido un importante deterioro y probablemente necesite el apoyo de otras áreas tales como geriatría, clínica del dolor y cuidados paliativos.
La mayoría de los pacientes en estas circunstancias están recibiendo terapia de mantenimiento al momento de la recaída, ya sea a base de IP o IMiD, por lo tanto, la recomendación en ellos es el inicio de combinaciones que incluyan al menos un fármaco que no hayan recibido previamente.
El carfilzomib, un IP de segunda generación, ha demostrado un buen perfil de seguridad y eficacia, en estudio fase II, en pacientes en recaída o con refractariedad al bortezomib y/o la lenalidomida, con tasas de respuesta global del 50% y supervivencia global a 12 meses del 60%, con perfiles de seguridad aceptables, por lo menos para pacientes en buen desempeño físico.7 En la tabla 4 se observa la eficacia encontrada en diversos estudios con inhibidores de proteosoma en este grupo de pacientes.
Tabla 4 Ensayos clínicos con esquemas basados en IP para pacientes con mieloma múltiple recaída/refractario
| Tratamiento | Tasas de respuesta % | RC % | SLP meses | SG meses |
|---|---|---|---|---|
| ENDEAVOR Kd vs. Vd Dimopoulos, et al., 20167 | 77 vs. 63 | 13 vs. 6 | 18.7 vs. 9.4 HR: 0.53 | NA vs. 24.3 HR: 0.79 |
| CASTOR DVd vs. Vd Palumbo, et al., 20169 | 84 vs. 63 | 29 vs. 10 | 16.7 vs. 7.1 HR: 0.31 | NA HR: 0.63 |
RC: respuesta completa; SLP: supervivencia libre de progresión; SG: supervivencia global; HR: hazard ratio; NA: No Aplica ; Kd: Carfilzomib-Dexametasona ; Vd: Bortezomib_Dexametasona ; DVd: Daratumumab-Bortezomib-Dexametasona.
El daratumumab, un anticuerpo monoclonal, fue inicialmente utilizado en pacientes en recaída con múltiples líneas de tratamiento previas en combinación con dexametasona, con tasas de respuesta global de hasta el 26%. Posterior a ello, demostró mejorar dichas tasas de respuesta al combinarlo con bortezomib o lenalidomida más dexametasona, con tasas de respuesta global de hasta un 93%. El perfil de seguridad de este medicamento es aceptable, a pesar de la alta tasa de reacciones relacionadas a la infusión (sin embargo, manejables), considerándose una buena opción en casos de refractariedad a IP o IMiD.8,9
La pomalidomida, un IMiD de tercera generación indicado en pacientes refractarios a la lenalidomida, ha demostrado ser eficaz tanto en aquellos en buen estado físico como en pacientes frágiles, esto demostrado en diversos estudios fase 2 de comparación de pomalidomida contra otros esquemas en pacientes incluso con resistencia a pomalidomida (Tabla 5). Su posología la convierte en una buena opción para pacientes que no pueden o no desean ser sometidos a administración de medicamentos intravenosos o subcutáneos.10
Tabla 5 Ensayos clínicos con esquemas basados en pomalidomida para pacientes con mieloma múltiple refractario a lenalidomida
| Tratamiento | Tasas de respuesta % | SLP meses | SG meses |
|---|---|---|---|
| Pom/Dex (n = 302) Richardson, et al., 201410 | 31 vs. 10 | 4.0 vs. 1.9 | 12.7 vs. 8.1 |
| Bortezomib + Pom/Dex (n = 559) Richardson, et al., 201911 | 82 vs. 50 | 11 vs. 7 | NA |
| Carfilzomib + Pom/Dex (n = 57) Shah, et al., 201512 | 62 | 10.3 | NA (1 año: 67%) |
| Daratumumab + Pom/Dex (n = 103) Chari, et al., 201713 | 60 | 8.8 | 17.5 |
| Ixazomib + Pom/Dex (n = 32) Krishnan, et al., 201814 | 48-58 | - | - |
SLP: supervivencia libre de progresión; SG: supervivencia global; NA: No Aplica; Pom/Dex: Pomalidomida/Dexametasona.
Modificado de Richardson PG et al.10, 11, Shah JJ. Et al.12, Chari A et al.13, Krishnan, et al., 201814.
La figura 4 muestra los esquemas recomendados en pacientes con recaída/refractariedad.

Módulo 5: Criterios de respuesta en mieloma múltiple
La medición de la enfermedad es indispensable al establecer el diagnóstico, al tercer ciclo y al término del protocolo, esto con evidencia del valor pronóstico demostrado por el grupo Pethema-GEM. Para dicha valoración se deberán realizar idealmente los estudios de la tabla 1, de manera comparativa con la evaluación inicial.
Tabla 1 Estudios de evaluación de respuesta a la inducción
| Estudio | Mensual | Trimestral | Semestral |
|---|---|---|---|
| Biometría hemática | + | + | + |
| Bioquímica, incluyendo calcio, creatinina y proteínas totales (albúmina, globulina) | + | + | + |
| Lactato deshidrogenasa | + | + | + |
| Electroforesis de proteínas séricas | + | + | |
| Recolección de orina de 24 h para electroforesis de proteínas | + | + | |
| Cuantificación de inmunoglobulinas (nefelometría) | + | + | |
| Inmunofijación en suero y orina | + | + | |
| Medición de cadenas ligeras séricas | + | + | |
| Estudio radiológico óseo que incluye columna vertebral, pelvis y cráneo | + | ||
| Aspirado y/o biopsia de médula ósea: morfología e inmunofenotipo | + | ||
| Citogenética de las células plasmáticas de la médula ósea (cariotipo metafásico y FISH) *semestral* | |||
| Gammagrama óseo, TC o RM si está clínicamente indicado, como en el caso de plasmocitoma al diagnóstico | + | ||
| PET-CT | + cuando sea posible. | ||
FISH: hibridación fluorescente in situ; TC: tomografía computarizada; RM: resonancia magnética; PET-CT: tomografía por emisión de positrones-tomografía computarizada.
Adaptada de Rajkumar, et al., 20141.
En la tabla 2 se muestra la categorización de la respuesta al tratamiento según los criterios del Grupo internacional de mieloma múltiple.
Tabla 2 Criterios de respuesta al tratamiento. Grupo internacional de mieloma múltiple
| RCe Respuesta completa estricta | RC + cadenas ligeras normales y ausencia de células clonales en citometría de flujo o por inmunohistoquímica en MO |
| RC Respuesta completa | Inmunofijación negativa en suero y orina, desaparición de plasmocitomas y menos del 5% de BMPC |
| MBRP Muy buena respuesta parcial | Componente monoclonal detectable en suero y orina por inmunofijación, pero no en la electroforesis o reducción menor del 90% en el componente sérico con componente monoclonal urinario menor de 100 mg/24 h |
| RP Respuesta parcial | Reducción del componente monoclonal sérico del 50% o mayor en orina del 90% o más, y/o < 200 mg/24 h, así como una reducción en el 50% o más de diámetro de plasmocitomas |
| EE Enfermedad estable | Pacientes que no cumplen con criterios de RC, MBRC, RP o enfermedad progresiva |
| Progresión | Requiere uno o más de los siguientes criterios de CRAB: - Aumento en más del 25% respecto al valor basal en: • Componente monoclonal sérico (el incremento absoluto debe ser ≥ 0.5 g/dl) y/o • Componente monoclonal urinario (el incremento absoluto debe ser ≥ a 200 mg/24 h) y/o • Solo en pacientes sin proteína monoclonal sérica y/o urinaria medible: diferencia entre FLC (el incremento absoluto debe ser > 10%) - Plasmocitosis medular: el aumento debe ser > 10% - Desarrollo de nuevas lesiones óseas o plasmocitomas, o aumento del tamaño de las lesiones óseas o plasmocitomas existentes - Desarrollo de hipercalcemia (calcio sérico corregido > 11.5 mg/dl o 2.65 mmol/l), que es atribuido al trastorno proliferativo plasmocitario por sí solo |
| Recaída clínica | Requiere uno o más de los siguientes indicadores directos de aumento de la enfermedad y/o disfunción orgánica (hallazgos CRAB). Se utiliza hasta la progresión: - Desarrollo de nuevos plasmocitomas o lesiones óseas - Aumento del tamaño de plasmocitomas o lesiones óseas existentes. Un aumento mayor al 50% (y al menos 1 cm) por mediciones seriadas - Hipercalcemia (> 11.5 mg/dl o 2.65 mmol/l) - Disminución de la Hb > 2 g/dl, aumento de la concentración de creatinina > 2 mg/dl |
| Recaída desde respuesta completas | Uno o más de los siguientes: - Reaparición en orina y/o suero del componente monoclonal medido por inmunofijación o electroforesis - Desarrollo de plasmocitosis medular > 5% - Aparición de otros signos de progresión: plasmocitomas nuevos, lesiones osteolíticas o hipercalcemia |
MO: médula ósea; MBRP: Muy Buena Respuesta Parcial; CRAB: hipercalcemia, insuficiencia renal, anemia y lesiones líticas; FLC: Free Light Chain; Hb: hemoglobina.
Adaptada de Munshi, et al., 20172.
Es importante destacar que cualquier tipo de respuesta debe mantenerse por un mínimo de seis semanas. El seguimiento de la enfermedad durante la terapia se sugiere de forma mensual, con realización de biometría hemática, calcio y proteínas totales. La cuantificación en suero y orina de proteína monoclonal se debe realizar de manera sistemática cada tres meses. En pacientes con mieloma de cadenas pesadas el seguimiento se realizará con electroforesis en orina y/o suero2.
Se pueden hacer recomendaciones para formas específicas de mieloma. En pacientes con cadenas pesadas de isotipo IgA con una espiga M en suero con migración electroforética en la región beta, otras proteínas séricas pueden confundir la evaluación precisa del componente monoclonal. En esos casos, la IgA total por nefelometría ha sido una herramienta útil para el seguimiento cuantitativo. La aparición de anticuerpos específicos contra IgA-kappa e IgA-lambda también se puede utilizar para discriminar entre IgA monoclonal y policlonal, lo que permite una medición más precisa. Cuando un paciente solo tiene cadena ligera monoclonal con proteína sérica monoclonal, pero la Inmunofijación es negativa para IgG, IgA o IgM, la posibilidad de IgD monoclonal se debe considerar. Se requiere cuantificación sérica de IgD y electroforesis en orina de 24 h en pacientes con mieloma IgD. Para casos de mieloma oligosecretor, componente M sérico menor de 10 g/l y componente de orina menor de 200 mg/24 h han sido establecido como criterios diagnósticos3,4.
En pacientes con sospecha de recaída se deberá iniciar el tratamiento solo si se tienen criterios clínicos o bioquímicos de recaída.
Módulo 6: Utilidad de la citometría de flujo en el diagnóstico de mieloma múltiple
Debido a la subjetividad de la morfología y a la imposibilidad de esta para demostrar clonalidad, la evolución de la citometría de flujo se ha posicionado en el manejo clínico integral del mieloma múltiple y otras alteraciones de las células plasmáticas, de forma sensible y específica, con la finalidad de caracterizar a las células plasmáticas, diferenciando fenotipos normales (Tabla 1) de los aberrantes por medio de paneles de anticuerpos que han sido estandarizados, lo que permite la comparación de estos entre diversos centros diagnósticos, unificando la información que se proporciona al médico tratante1. La única estrategia estandarizada es la diseñada por el consorcio EuroFlow, que publicó los paneles para el diagnóstico y pronóstico (Tabla 2), y para el monitoreo de enfermedad mínima residual (EMR) (Tabla 3).
Tabla 1 Fenotipo habitual de células plasmáticas normales
| Marcador | Expresión |
|---|---|
| CD38 | ++ |
| CD138 | + |
| CD19 | + |
| CD45 | + |
| CD56 | - |
| cyIgk/cyIgl | policlonal |
| b2-microglobulina | + |
| CD81 | + |
| CD117 | |
| CD27 | + |
| CD28 | |
+: expresión positiva; ++: expresión positiva intensa; : expresión negativa.
Tabla 2 Panel EuroFlow para el diagnóstico en células plasmáticas
| Tubos | Marcadores y fluorocromos | |||||||
|---|---|---|---|---|---|---|---|---|
| PacB | PacO | FITC | PE | PerCP-Cy 5.5 | PE-Cy 7 | APC | APC-H7 | |
| Tubo 1 Diagnóstico y clonalidad | CD45 | CD138 | CD38 | CD56 | b2-microglobulina | CD19 | cyIgk | cyIgk |
| Tubo 2 Pronóstico | CD45 | CD138 | CD38 | CD28 | CD27 | CD19 | CD117 | CD81 |
PacO: pacific orange; PacO: pacific blue; PE: ficoeritrina; PE-Cy 7: ficocianin cyanin 7; PerCP-Cy 5.5: peridinin chlorophyll-protein-cyanin 5.5; APC: aloficocianina; FITC: isotiocianato de fluoresceína; APC-H7: aloficocianina H7.
Adaptada de Kalina, et al., 20122.
Tabla 3 Panel EuroFlow para el monitoreo de enfermedad mínima residual
| Tubos | Marcadores y fluorocromos | |||||||
|---|---|---|---|---|---|---|---|---|
| BV421 | BV510 | FITC | PE | PerCP- Cy 5.5 | PE-Cy 7 | APC | APC-H7 | |
| Tubo 1 | CD138 | CD27 | CD38* | CD56 | CD45 | CD19 | CD117 | CD81 |
| Tubo 2 | CD138 | CD27 | CD38* | CD56 | CD45 | CD19 | cyIgk | cyIgk |
*Opcional cambio a CD38-multiepitope en paciente con terapia anti-CD38.
BV421: brilliant violet 421; BV510: brilliant violet 510; PE: ficoeritrina; PE-Cy7: ficocianin cyanin 7; PerCP-Cy 5.5: peridinin chlorophyll-protein-cyanin 5.5; APC: aloficocianina;
FITC: isotiocianato de fluoresceína; APC-H7: aloficocianina H7.
Adaptada de Flores-Montero, et al., 20173.
Para poder discriminar las alteraciones fenotípicas en las células plasmáticas aberrantes es imprescindible conocer la utilidad antigénica que define, la cual se describe en la tabla 4.
Tabla 4 Utilidad de los marcadores para células plasmáticas
| TUBO | Antígenos | Identificación de células plasmáticas | Marcadores aberrantes | Identificación de clonalidad |
|---|---|---|---|---|
| Marcadores comunes | CD38 CD138 CD19 CD45 |
X X X X |
C C C |
|
| Tubo 1 | cyIgl cyIgk CD56 β2-microglobulina |
C P |
X X |
|
| Tubo 2 | CD27 CD28 CD117 CD81 |
P P P P |
C: marcadores aberrantes comunes; P: marcadores pronósticos.
Adaptada de Kalina, et al., 20122.
Las especificaciones recomendadas por el National Comprehensive Cancer Network del 2019 para realizar citometría de nueva generación en el monitoreo de la EMR son las siguientes:
1) Sensibilidad: 1 en 100,000 eventos (10-5), adquisición de al menos 10 millones de células (107).
2) Utilizar los procedimientos de operación estandarizados por EuroFlow para EMR en MM3,4.
Con respecto a las especificaciones del monitoreo de la EMR en el MM, de acuerdo con la reunión de expertos en 2018 se recomienda realizarla en todos los pacientes que logren una respuesta completa o muy buena respuesta parcial, de acuerdo con los criterios del Grupo Internacional de trabajo de Mieloma5.
Módulo 7: Trasplante de células progenitoras hematopoyéticas en mieloma múltiple
El mieloma múltiple (MM) es una enfermedad incurable que compone el 10-13% de las neoplasias hematológicas. La mediana de edad al diagnóstico es de 65-70 años. El trabajo de Powles, Barlogie y McElwain condujo, 30 años después, al inicio del concepto de quimioterapia intensiva, o a dosis alta (HDT), seguido de un trasplante autólogo de células progenitoras hematopoyéticas (TCPH-A). El trasplante sigue siendo el estándar para el tratamiento del MM de reciente diagnóstico (NDMM) en pacientes jóvenes y ancianos con adecuada funcionalidad. La superioridad del procedimiento fue aprobada inicialmente por el Intergroupe Francophone du Myelome (IFM) y luego confirmada por el Consejo de Investigación Médica del Reino Unido. A pesar de su impacto significativo confirmado en la supervivencia libre de eventos (SLE), varios ensayos no lograron mostrar un impacto significativo en la supervivencia global (SG). Aunque considerado una terapia estándar, el procedimiento todavía se ve desafiado por recaídas inevitables que amenazan las remisiones prolongadas1.
Se ha demostrado que el TCPH-A temprano es superior al tardío (en la recaída), incluso en la era de los nuevos agentes. En un metaanálisis en el cual se analizaron 3,829 pacientes con MM de novo que recibieron altas dosis de quimioterapia con TCPH-A temprano comparado con quimioterapia estándar o trasplante tardío, los resultados fueron favorables para el trasplante temprano con SLP con hazard ratio (HR) de 0.73 (intervalo de confianza [IC] 95%: 0.56-0.94), aunque esta diferencia no se observó en SG, con un HR de 0.86 (IC 95%: 0.70-1.04)1.
La mortalidad relacionada al trasplante (MRT) con TCPH-A es muy baja (1-2%), la mediana de SLP es de 50-56 meses y la mediana de SG esperada es de 8-10 años. Mediante un análisis multivariante se observó que la respuesta completa (RC) estricta es un factor pronóstico independiente para mejores supervivencias (HR: 0.44 ; IC 95%: 0.25-0.80)2.
Indicaciones del trasplante de células progenitoras hematopoyéticas en el mieloma múltiple
El TCPH-A en MM sigue siendo el tratamiento de consolidación de elección en aquellos pacientes elegibles para trasplante (Figura 1). Desde la década de los 90 se demostraron mejores tasas de respuesta global, profundizando incluso la tasa de RC entre un 15-20%. Igualmente, se han conseguido mejores tasas de SG y SLP en los pacientes sometidos a TCPH. En estudios subsecuentes, el mayor beneficio del trasplante se ha observado en términos de SLP, siendo más inconsistentes en demostrar este mismo beneficio en términos de SG.

Figura 1 Algoritmo de tratamiento de Mieloma múltiple.TCH: trasplante de células hematopoyéticas; TC: tomografía computarizada; FDG-PET/CT: tomografía por emisión de positrones/tomografía computarizada con 18F-fluorodesoxiglucosa; RM: resonancia magnética.
Este beneficio se había demostrado con los esquemas de inducción basados en quimioterapia citotóxica, sin embargo, el beneficio en la era de las terapias novedosas (inmunomoduladores, inhibidores del proteosoma, etc.) con las que se consiguen mejores desenlaces globales no estaba tan claro1. Recientemente se han publicado los resultados de un ensayo clínico fase III que evaluó el beneficio del TCPH en el escenario de terapias actuales (RVD vs. RVD-TCPH), demostrando beneficio con los pacientes sometidos a TCPH en SLP (50 vs. 36 meses; p < 0.001) en todos los subgrupos de pacientes. Igualmente, se observó una mejor RC (59 vs. 48%; p = 0.03) y negativización de enfermedad mínima residual (EMR) (79 vs. 65%; p < 0.001), sin impacto en SG a cuatro años ni en MRT3.
Elegibilidad
No existe consenso sobre un límite de edad más allá del cual el tratamiento con TCPH se vuelva cuestionable y, como tal, la práctica varía según institución y país.
La edad avanzada ya no es un criterio de exclusión para determinar la elegibilidad a trasplante. En un ensayo fase II no existió diferencia en la MRT en pacientes 60 a 65 años vs. 65 a 70 años, con baja tasa de MRT en las dos cohortes (< 1%)4.
Un estudio previo en el que la mediana de edad de los pacientes era de 72 años concluyó que los pacientes con MM de edad avanzada no deberían ser excluidos del trasplante, mostrando buenos resultados con melfalán 140 mg/m2.
En un consenso realizado por la Mayo Clinic recomendaron que, en contraste con los estrictos límites de edad cronológicos para la elegibilidad para TCPH, el estado de funcional y las comorbilidades deben considerarse para la elegibilidad para TCPH. Recomiendan, además, que la profundidad de la quimioterapia de inducción no debe dictar la elegibilidad a un trasplante. Ambas recomendaciones con un nivel de evidencia III, grado de recomendación B.
La indicación de TCPH-A en NDMM o en 1.a recaída debe ser extendida a todo paciente con respuesta parcial o mejor, independientemente del grado de profundidad alcanzada durante inducción. No es necesario evaluar la EMR de manera sistemática, sin embargo, se recomienda evaluar en todo aquel paciente que se encuentra en respuesta completa estricta previa al trasplante para documentar la profundidad de la respuesta, toda vez que se ha demostrado que la respuesta previa al trasplante es un factor determinante para la SLP en estos pacientes.5
Escalas de comorbilidad
Algunos autores recomiendan el uso de escalas de comorbilidad para valorar la elegibilidad de un paciente para recibir un TCPH. Las tres escalas más usadas son el HCT Comorbidity Index (HCT-CI), el Charlson Comorbidity Index (CCI) y el sistema de puntuación Pretransplant Assessment of Mortality (PAM); este último más usado para los trasplantes alogénicos5 (Tabla 1).
Tabla 1 Comparación del índice de comorbilidad de Charlson (CCI) y del índice de comorbilidad específico de trasplante de células hematopoyéticas (HCT-CI)
| Comorbilidad | Definición | CCI score | HCT-CI definiciones | HCT-CI puntaje |
|---|---|---|---|---|
|
Enfermedad pulmonar
Leve Moderada Grave |
Disnea en actividad moderada Disnea con actividad ligera Disnea en reposo con oxígeno |
1 1 1 |
Disnea con moderada actividad o DLco y/o FEV1 81-90%
Disnea con ligera actividad DLco y/o FEV1 66 a 88% Disnea en reposo o requiere oxígeno o DLco y/0 FEV1 ≤ 65% |
0 2 3 |
|
Enfermedad cardiaca
General Arritmia Enfermedad valvular |
Falla cardiaca congestiva N/A Excepto prolapso de la válvula mitral |
1-2 0 0 |
Enfermedad coronaria, insuficiencia cardiaca
congestiva, IAM, FEVI < 50% Fibrilación auricular o flutter, seno enfermo, arritmias ventriculares Excepto prolapso de la válvula mitral |
1 1 3 |
|
Enfermedad hepática
Leve Moderada a grave |
] Hepatitis crónica o cirrosis Hipertensión portal, varices esofágicas |
1 3 |
Hepatitis crónica, bilirrubina > 1.5 veces o AST/ALT >
2.5 veces Cirrosis o fibrosis o bilirrubina > 1.5 veces o AST/ALT > 2.5 veces |
1 3 |
|
Enfermedad renal Leve Moderada a grave |
Creatinina 2-3 mg/dl Creatinina > 3, diálisis, trasplante renal |
0 2 |
Creatinina 1.2-2 mg/dl Creatinina > 2 mg/dl, diálisis o trasplante renal |
0 2 |
|
Otras neoplasias
Tumor sólido Cáncer metastásico |
Tratado en los últimos 5 años Presente |
2 6 | Tratado en cualquier tiempo de la historia clínica del
paciente No incluido |
1 N/A |
|
Otras comorbilidades
Diabetes Enfermedad cerebrovascular Reumatológica Ulcera péptica |
Tratamiento con insulina y/o hipoglucemiantes orales
Evento vascular, ataque isquémico transitorio LES, AR, polimiositis, enfermedad mixta de tejido conectivo Requiere tratamiento |
1 1 1 1 |
Tratamiento con insulina y/o hipoglucemiantes orales
Evento vascular, ataque isquémico transitorio LES, AR, polimiositis, enfermedad mixta de tejido conectivo Requiere tratamiento |
1 1 2 2 |
N/A: No Aplica; LES: lupus eritematoso sistémico; AR: artritis reumatoide; Dlco: capacidad de difusión de monóxido de carbono; FEV1: volumen espiratorio forzado el primer segundo; IAM: infarto agudo de miocardio; AST: aspartato aminotransferasa; ALT: alanina aminotransferasa.
N/A: No Aplica; LES: lupus eritematoso sistémico; AR: artritis reumatoide; Dlco: capacidad de difusión de monóxido de carbono; FEV1: volumen espiratorio forzado el primer segundo; IAM: infarto agudo de miocardio; AST: aspartato aminotransferasa; ALT: alanina aminotransferasa.
Aunque no existen en sí criterios definidos para la indicación del trasplante en el paciente con MM, existen cada vez más estudios que reflejan que una remisión completa debería ser un objetivo importante en el paciente al momento de pensar en un trasplante. La profundidad de respuesta (idealmente la EMR negativa medular y extramedular) se correlaciona con mayor SLP y SG en todos los subgrupos de pacientes, incluidos los de alto riesgo citogenético.
En un consenso realizado por el International Myeloma Working Group en el 2016 se dio como recomendación iniciar HDT con TCPH como la terapia estándar para pacientes elegibles para trasplante con NDMM. Contribuye a mejorar el resultado en todos los grupos de pronóstico. La HDT/TCPH doble combinada con bortezomib puede mejorar la SLP en pacientes con t (4;14) o del (17p) y en aquellos con ambas anormalidades. Aunque los resultados de los ensayos aleatorios estratificados aún no están disponibles, se recomienda HDT más TCPH-A doble para pacientes con citogenética de alto riesgo.6
La realización del trasplante de manera temprana vs. tardía se ha analizado en varios estudios. En la era de la quimioterapia convencional, ya desde 1998 el grupo francés reportó los resultados en ambos brazos, siendo comparables en SG; sin embargo, el trasplante temprano fue superior en términos de calidad de vida al reducir la toxicidad asociada a tratamiento y aumentando el tiempo sin síntomas y/o tratamiento. En los ensayos más recientes es evidente el beneficio del trasplante al profundizar la respuesta al tratamiento y aumentar la SLP. Con base en estos datos, es seguro concluir que el TCPH-A mejora significativamente los desenlaces globales tanto en primera línea como en el escenario de rescate, por lo que debe seguir siendo parte del esquema de consolidación en pacientes elegibles a trasplante con NDMM (1.a línea)7.
En la era de la quimioterapia convencional, el TCPH en tándem era una opción terapéutica que mejoraba los desenlaces globales en términos de SLP y SG, incluso en pacientes que no lograban RC previa a trasplante1. En la era de la inmunoterapia, se requiere reevaluar el papel del trasplante en tándem. Recientemente se han publicado los resultados del estudio EMN02/HO95 (RVD) donde se demostró impacto benéfico significativo en términos de RC, SG y SLP en el brazo del trasplante en tándem, incluso en el subgrupo de alto riesgo citogenético. Por otra parte, en el estudio StaMINA no se logró identificar este mismo beneficio1.
El TCPH alogénico en MM es controvertido. Los primeros estudios fallaron en demostrar un beneficio clínico por el aumento en la MRT. Posteriormente, todavía en la era de los esquemas de inducción basados en quimioterapia citotóxica, se propuso el trasplante en tándem (auto-TCPH seguido de alo-TCPH de intensidad reducida), consiguiendo resultados discordantes en los diferentes estudios, probablemente por la heterogeneidad en los esquemas de acondicionamiento, profilaxis para la enfermedad de injerto contra huésped y diseño de los ensayos clínicos. En la era de las terapias novedosas, el papel del trasplante alogénico tiene lugar en el escenario del paciente con recaída tras auto-TCPH y dentro del escenario de ensayos clínicos, sobre todo por la modesta actividad de injerto contra tumor en el MM7.
Movilización
El TCPH-A en el MM se enfrenta al problema de alcanzar una dosis celular adecuada en la cosecha para este, siendo una de las enfermedades consideradas como pobres movilizadores; el problema se incrementa cuando se pretende cosechar para trasplante tándem. La cantidad mínima de células progenitoras de sangre periférica (PBSC) requerida para un TCPH-A es 2x106 células CD34+/kg; cuando se tiene una cosecha < 1x106/kg se asocia a un injerto tardío con altos requerimientos transfusionales e incluso pérdida permanente de injerto, mientras que 3-5x106 células CD34+/kg son ideales8.
Hay dos estrategias diferentes para movilizar células progenitoras hematopoyéticas (CPH) de la médula ósea a la sangre periférica (SP): a) factores estimulantes de colonias de granulocitos (G-CSF), y b) secuencial con quimioterapia seguida de G-CSF.
Estimulante de colonias
Para la movilización con G-CSF los únicos aprobados son el filgrastim y el lenograstim. Las dosis recomendadas son filgrastim 10 µg/kg/día subcutáneo (SC) durante 5-7 días consecutivos y lenograstim 10 µg/kg/día SC durante 4-6 días consecutivos. El uso de un G-CSF biosimilar tiene una eficacia equivalente.9 El uso de pegfilgrastim de 6 y 12 mg SC, dosis única, también tiene evidencia favorable en cosecha para autotrasplante; en un metaanálisis se comparó pegfilgrastim vs. filgrastim, demostrando mayor cosecha de CD34 y día de cosecha más temprano para los pacientes con pegfilgrastim. No se encontró diferencia para el número de aféresis realizadas ni en el día de injerto al trasplante.
Se espera lograr en la primera movilización el número de CD34+ ideal, con la menor toxicidad, por lo que se monitoriza la cifra de CD34 en SP para escoger el primer día de aféresis, siendo más tempranamente cuando se usa pegfilgrastim entre el día 3-5, para filgrastim del día 5-7 y para quimioterapia + G-CSF entre el día 10-13 posquimioterapia. El día ideal de inicio de cosecha es donde se espere tener > 10 X 1010 células CD34+.10
Si el número de células recolectadas es inadecuado, la movilización con G-CSF puede continuar durante 1 a 2 días. Sin embargo, si no se alcanza el objetivo de recolección después de la tercera leucoféresis, es improbable una movilización exitosa.
Las principales ventajas de la movilización con G-CSF son la toxicidad relativamente baja, el tiempo predecible de leucoféresis, la administración ambulatoria y los costos reducidos en comparación con la quimiomovilización. Las principales desventajas son las tasas variables de fracaso de la movilización y los menores rendimientos de células CD34+ en comparación con la quimiomovilización. La movilización con G-CSF solo puede usarse en pacientes sin necesidad adicional de quimioterapia, por ejemplo, en pacientes con una remisión estable de la enfermedad subyacente. Se estima que alrededor del 20% de los MM no podrán alcanzar una cosecha adecuada bajo esquema de G-CSF.
Movilización con quimioterapia
El uso de quimioterapia en combinación con G-CSF es la forma preferida de movilización para todos los pacientes que necesitarán una mayor disminución de la carga tumoral y/o que tengan que recolectar una gran cantidad de CPH.
La dosis más utilizada es ciclofosfamida (CY) 2-4 g/m2, también llamado quimiomovilización, que ha demostrado proporcionar mayor número de CD34+ para la cosecha. Dosis mayores de CY se han asociado con neutropenias prolongadas, uso de antibióticos y transfusiones.
Otro esquema que ha demostrado efectividad son dosis intermedias de citarabina (Ara-C) con dosis de 1.6 g/m2 + G-CSF como esquema de movilización de primera o segunda línea, que en un estudio retrospectivo demostró ser más eficiente que CY + G-CSF o G-CSF solo, con una mediana de aféresis de un día comparado con dos con G-CSF solo con mediana de cosecha de 20.2 (2.9-59.4) x106 CD34+/kg con Ara-C vs. 5.9 (0-11) x106 CD34+/kg (p < 0.00001). La toxicidad fue mayor en el grupo que recibió quimioterapia con neutropenia grado 3/4 en el 34% y trombocitopenia 3/4 en el 75%11.
Se cuenta, además, con alta efectividad en movilización con plerixafor, antagonista de CXC-4, en combinación con G-CSF, con el cual se ha demostrado que se logra cosechar > 2x106 CD34/kg en la primera recolección (odds ratio [OR]:4.05; IC 95%: 1.19-13.83; p=0.03) y ≥ 5x106/kg CD34+ en total de cosecha (OR:3.09; IC 95%: 1.04-9.23; p=0.04)12.
Predictores de movilización subóptima
El fracaso de la movilización se define generalmente como la incapacidad de obtener 2 × 106 células CD34+/kg en cuatro sesiones de aféresis. A pesar de los avances recientes en las estrategias de recolección de PBSC, la imposibilidad de obtener una dosis celular adecuada continúa retrasando y excluyendo el auto-HCT en candidatos a trasplantes adecuados. Los factores asociados con la movilización inadecuada de CPH en pacientes con MM incluyen: trombocitopenia, edad > 60 años, curso de tratamiento extenso, radioterapia previa, exposición previa a agentes alquilantes y uso prolongado de lenalidomida13 (Figura 2).

Acondicionamiento
Esquema de acondicionamiento
En pacientes con MM sometidos a TCPH-A, el acondicionamiento estándar es melfalán 200 mg/m2. Se han realizado múltiples estudios con el objetivo de buscar el mejor esquema de acondicionamiento.
En 2017 el IFM realizó un estudio abierto, aleatorizado, fase III, donde comparó melfalán 200 mg/m2 vs. bortezomib 1 mg/m2 por 4 días + melfalán 200 mg/m2 1 día; no se encontró diferencia en las respuestas, la SG ni eventos adversos. Sin embargo, el esquema no ha sido evaluado respecto a EMR14.
En otro estudio fase II que comparó busulfano 130 mg/m2/día por 4 días + melfalán 70 mg/m2 por 2 día, se observó un incremento en la SLP, pero sin diferencia en las tasas de respuesta. En 2019, Bashir, et al., en un estudio fase II con 202 pacientes compararon busulfano 32 mg por 4 días + melfalán 140 mg/m2 vs. melfalán 200 mg/m2, observando una SLE de 64.7 vs. 43.5 meses, respectivamente (HR: 0.53; IC 95%: 0.30-0.91; p = 0.022). Se reportaron más eventos adversos no hematológicos grados 3 y 4, principalmente diarrea y mucositis, pero menor incidencia de enfermedad venooclusiva hepática.15
Otro esquema evaluó el uso de bortezomib (1.3 mg/m2 adicionado a BuMel [busulfano y clorhidrato de melfalán]) en un estudio fase II. Se encontró un 75% de muy buena respuesta parcial o mejor, un 55% de RC a los tres meses tras el TCPH-A; sin embargo, serán necesario estudios fase III para poder comparar entre Bortezomib-BuMel vs. BuMel vs. melfalán 20016.
La adición de bendamustina 200 mg/m2 por 2 días + melfalán 100 mg/m2 por 2 días está siendo explorada con resultados prometedores en las tasas de respuesta y SLP en el segundo trasplante con citogenética desfavorable17. Recientemente se propuso un algoritmo de procedimiento terapéutico y trasplante de acuerdo al riesgo inicial, donde resalta el trasplante de células progenitoras hematopoyéticas en los pacientes de riesgo intermedio y alto inmediatamente después de la inducción y posterior al trasplante no olvidar el mantenimiento, como se puede observar en la figura 3.

Figura 3 Esquema de terapia de inducción.*Si > 70 años o CrCl < 30 ml/min, considerar Mel 140 mg/m2.TCPH-A: trasplante autólogo de células progenitoras hematopoyéticas; Mel: melfalán; CrCl: aclaramiento de creatinina.Adaptada de Gonsalves, et al., 2019.5
Casos especiales
En el caso de la enfermedad extramedular, se ha evaluado en estudios retrospectivos el uso del esquema de acondicionamiento BEAM (dexametasona con carmustina, etopósido, Ara-C y melfalán), que se usa típicamente como acondicionamiento en pacientes con linfoma y puede ser efectivo18.
En pacientes con enfermedad renal al diagnóstico (tasa de filtrado glomerular < 30 ml/min o creatinina > 2 mg/dl), se evaluó de forma retrospectiva la utilización de melfalán 140 mg/m2 vs. melfalán 200 mg/m2; La dosis de 140 mg fue menos tóxico sin diferencias en mortalidad asociada a trasplante, SLP y SG. Respecto a los pacientes mayores de 70 años en buenas condiciones, no deben ser excluidos, debido a que el acondicionamiento con melfalán 140 mg/m2 es menos tóxico e igual de eficaz que con melfalán 200 mg/m2,19.
Hasta el momento, el consenso Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) corroboró que el melfalán 200 mg/m2 sigue siendo la terapia de acondicionamiento estándar utilizada fuera de un ensayo clínico. Se sugiere la participación en ensayos clínicos con nuevos agentes como esquemas de acondicionamiento para continuar en la búsqueda del mejor esquema de acondicionamiento. Nivel de evidencia I, grado de recomendación A5 (Figura 3).
Mantenimiento postrasplante
El objetivo debe ser prolongar la duración de la remisión sin afectar la calidad de vida. La talidomida produce un 7-19% de polineuropatía periférica grado 3-4, lo que lleva a la suspensión de esta, por lo que no es un medicamento ideal.
El bortezomib (estudio HOVON-65/GMMG-HD4) reduce significativamente el riesgo de progresión (p = 0.04) y muerte (p = 0.05) comparado con la talidomida, pero con tasa similar de PNP (5 vs. 8%). Se sugiere que la inducción pretrasplante y mantenimiento con este reduce significativamente la supervivencia en pacientes con del (17p) y recaída.
La lenalidomida es menos neurotóxica (cuatro estudios demostraron beneficio en SLP de mantenimiento con lenalidomida vs. no darlo (HR: 0.46-0.5) y estudios aleatorizados reportan un incremento de SG (SG 7 años, 62 vs. 50%; HR: 0.75; p = 0.001), excepto en pacientes con alto riesgo citogenético, y mejora la SLP. La principal toxicidad grado 3-4 es neutropenia (23 vs. 51%) e infecciones (6-13%), aunque las segundas neoplasias son mayores (hematológicas 6.1 vs. 2.8% y tumores sólidos 7.3 vs. 4.2%)20.
En el estudio TOURMALINE-MM3 se evaluó el mantenimiento postrasplante con ixazomib contra placebo. Se observó una reducción del 28% en el riesgo de progresión o muerte, en la SLP (26.5 meses [IC 95%: 23.7-33.8] vs. 21.3 meses [IC 95%: 18-24.7]; OR: 0.72 [IC 95%: 0.58-0.89].21
El carfilzomib es seguro como mantenimiento por un año en NDMM (incluyendo mayores de 75 años) y lleva a una respuesta más profunda (50% de RC al final del mantenimiento, con una SLP a 28.1 meses y SG estimada a 36 meses de aproximadamente el 70%; p = 0.0023). No hubo incremento en segundas neoplasias con ixazomib: 12 (3%) pacientes, contra 8 (3%) pacientes con placebo; 108 (27%) de 394 pacientes en el grupo de ixazomib y 51 (20%) de 259 pacientes en el grupo placebo, experimentaron eventos adversos. Durante el tratamiento murió un paciente en el grupo de ixazomib y ninguno en el grupo placebo21.
Un resumen de las características de ambos esquemas de acondicionamiento se presenta en la tabla 2.
Tabla 2 Esquemas de mantenimiento post trasplante
| Mantenimiento (Nivel de evidencia) | Tratamiento de elección | |
|---|---|---|
| Lenalidomida (IA) Talidomida (IA) Bortezomib (IB) | - Lenalidomida: tiene mejor perfil de toxicidad que la
talidomida, lo que favorece su uso a largo plazo - Bortezomib: es más seguro y eficaz que la talidomida - IMiD: solos podrían ser subóptimos en pacientes de alto riesgo y en pacientes con insuficiencia renal, quienes podrían beneficiarse del bortezomib - Ixazomib: reduce el riesgo de progresión o muerte comparado con placebo - Carfilzomib: es seguro y eficaz |
- El tratamiento de elección debe considerar las características del paciente y la toxicidad esperada |
| Duración de la terapia | ||
| - Lenalidomida: al menos dos años o hasta la tolerancia
- Talidomida: hasta la tolerancia - Bortezomib: dos años - Ixazomib: semanal por dos años - Carfilzomib: semanal por un año |
- No hay estudios aleatorizados que comparen
lenalidomida por 2 años con lenalidomida hasta la progresión
- El uso de talidomida a largo plazo es limitado por pobre tolerancia - El bortezomib ha sido administrado en ensayos clínicos hasta por dos años |
|
IMiD: inmunomoduladores.
Módulo 8: Tratamiento del dolor y soporte
El mieloma múltiple (MM) es una enfermedad que desde etapas tempranas se manifiesta con síntomas que interfieren con la calidad de vida.
Es importante mencionar que un paciente con MM tendrá afectadas las esferas física, social, emocional y espiritual, por lo que debe llevarse a cabo un manejo transdisciplinario.
La quimioterapia y la radioterapia a menudo pueden conllevar algunas complicaciones, sin embargo, se deben implementar tratamientos adicionales que mejoren el confort y la calidad de vida.
Dentro de los principales síntomas que presentan los pacientes se encuentra el dolor óseo, la neuropatía, la ansiedad, la fatiga y el insomnio, entre otros1.
Complicaciones óseas
La enfermedad ósea relacionada con mieloma (EOM) es una complicación frecuente que se presenta en diversos grados, desde la osteopenia leve; se estima que esta se presenta en el 85% de los casos y puede llegar hasta lesiones óseas severas que traen como consecuencia fracturas patológicas y sus complicaciones específicas. Esta enfermedad se debe a la pérdida del equilibrio en la reabsorción ósea mediada por los osteoclastos y la formación ósea, responsabilidad de los osteoblastos. Se sabe que las células del mieloma estimulan a los osteoclastos debido a la liberación aumentada de interleucinas, factor estimulante de colonias de macrófagos, factor de necrosis tumoral (TNF) alfa y beta, y otras moléculas que a su vez estimulan al osteoclasto a liberar diversas moléculas que estimulan la replicación de células de MM, cerrando así el círculo vicioso2.
Últimamente se le ha dado gran importancia a una vía de señalización que ha demostrado estar implicada en la enfermedad ósea de diversas patologías neoplásicas, incluyendo al mieloma: la vía del RANK-RANKL-OPG. Los osteoclastos derivan de precursores hematopoyéticos, la RANK (una proteína de la familia de las citocinas TNF que regula la replicación, movilización y función de estas células) y su ligando RANKL, regulado por la osteoprotegerina (OPG), secretada por las células estromales. Un desequilibrio en la relación RANKL/OPG tiene como consecuencia la función inapropiada de los osteoclastos y la reabsorción ósea alterada2.
Las lesiones de la EOM son de tipo lítico, se pueden presentar en cualquier hueso y están relacionadas con alta actividad tumoral. El diagnóstico y determinación de estas lesiones se ha hecho tradicionalmente con serie ósea metastásica, que, si bien es el estudio más barato y disponible en la mayoría de los centros médicos, tiene la gran desventaja de que solo es sensible a lesiones altamente avanzadas, considerándose que entre el 30 y el 50% del hueso puede sufrir de destrucción de forma previa a ser detectado por esta técnica3. La tomografía computarizada de baja dosis es actualmente uno de los estudios más ampliamente utilizados; su sensibilidad y especificidad, así como la relativa disponibilidad del equipo necesario para su realización, la convierten en una de las mejores opciones diagnósticas para esta complicación4. La resonancia magnética y la tomografía por emisión de positrones-tomografía computarizada son estudios con alta utilidad para la detección temprana de lesiones líticas, con alta sensibilidad; sin embargo, su disponibilidad en la mayoría de los centros médicos es nula y requieren de preparación más compleja por parte del paciente para su realización.
Tratamiento de la enfermedad ósea relacionada con mieloma múltiple
El tratamiento de la EOM se ha terminado posterior a la aprobación de los agentes utilizados para complicaciones óseas en otras neoplasias. La piedra angular del manejo son los bifosfonatos: análogos del pirofosfato que a su vez inhiben la pirofosfato sintasa, inhibiendo la proliferación y maduración de los osteoclastos, induciendo su apoptosis y evitando su fijación al hueso. El clodronato, el pamidronato y el ácido zoledrónico son los bifosfonatos actualmente aprobados para su uso en MM. Se han llevado a cabo diversos estudios fase III comparando la actividad y beneficio de estos medicamentos cara a cara. Uno de los más recientes y amplios es el realizado por el Medical research Council of the United Kingdom, llamado Myeloma IX Trial, en el cual se comparó el uso de clodronato frente a ácido zoledrónico, demostrando ser este último más eficaz en la disminución de las fracturas relacionadas con lesiones líticas, así como mejoría en la calidad de vida e incluso mejoría en la supervivencia global. La posología de ácido zoledrónico varía de acuerdo con el grupo consultado, sin embargo, la más comúnmente utilizada es 4 mg intravenoso cada cuatro semanas por un periodo de dos años5.
El perfil de seguridad de los bifosfonatos es aceptable, pero se deben considerar primordialmente dos eventos de gran importancia:
La osteonecrosis mandibular, que se puede presentar en entre el 2 y el 10% de los pacientes con uso de bifosfonatos. Esta complicación se caracteriza por la exposición de tejido óseo en cavidad oral. Entre los factores de riesgo para desarrollarla se encuentran: lesiones dentales previas al uso del medicamento, mala higiene dental, edad avanzada y evolución tardía del mieloma. Al presentarse esta complicación se debe interconsultar a expertos en el área de estomatología, y previo a ello se debe orientar al paciente a llevar una adecuada higiene dental, evitar procedimientos odontológicos innecesarios y vigilar la cavidad oral; en caso de requerir alguna intervención estomatológica quirúrgica urgente, se debe de suspender el uso del bifosfonato elegido durante hasta 90 días previos al procedimiento. Si ya se ha instaurado la complicación se debe iniciar tratamiento antibiótico de amplio espectro y consultar a los expertos6.
La insuficiencia renal aguda. Se puede ver en hasta el 12% de los pacientes con mieloma y uso de ácido zoledrónico, pues este se elimina en hasta el 40% por vía renal. No hay estudios del uso de este fármaco en pacientes con insuficiencia renal grave, sin embargo, la recomendación es la disminución y ajuste de la dosis del medicamento en caso de insuficiencia renal.
La recientemente descrita vía RANKL-OPG trajo consigo una nueva gama de oportunidades al encontrar nuevos blancos terapéuticos. Uno de los primeros desarrollos enfocados en esta área fue el denosumab, un anticuerpo monoclonal humano que se une e inhibe al RANKL de manera irreversible. Su eficacia se demostró en un principio en enfermedad metastásica ósea con tumores sólidos, para después ser analizado en la EOM, donde demostró no ser inferior al ácido zoledrónico en la supervivencia libre de evento (SLE), definida como fractura patológica o la necesidad de radioterapia paliativa (SLE de 22 vs. 23 meses), y mostró SG similares. Con respecto a la seguridad, sí se observó un ligero aumento en la frecuencia de osteonecrosis mandibular en el grupo de denosumab vs. el de ácido zoledrónico, pero sin diferencia estadísticamente significativa. La posología de este anticuerpo monoclonal es subcutánea, con dosis de carga los días 8 y 15 del primer ciclo y posteriormente 120 mg cada mes, lo que le confiere un buen perfil de practicidad y seguridad para el paciente2. Las ventajas del denosumab vs. el ácido zoledrónico son varias2:
‒ Administración subcutánea mensual.
‒ No se acumulan el hueso.
‒ Vida media de 26 días.
‒ Su depuración es a través del sistema reticuloendotelial (no es necesario ajustar la dosis de acuerdo con la función renal).
Las desventajas son:
En la tabla 1 se resumen las dosis y ajustes necesarios del ácido zolendrónico de acuerdo a caracteriticas de nuestros pacientes.
Tabla 1 Dosis de ácido zoledrónico
| - Dosis habitual 4 mg |
| - No se recomienda administración si CrCl < 30 ml/minuto |
| - Pacientes con IR leve a moderada (CrCl 30-60 ml/minuto) se recomienda: |
| • CrCl > 60 ml/minuto ® 4 mg |
| • CrCl 50-60 ml/minuto ® 3.5 mg |
| • CrCl 40-49 ml/minuto ® 3.3 mg |
| • CrCl 30-39 ml/minuto ® 3.0 mg |
CrCl: aclaramiento de creatinina; IR: insuficiencia renal.
La radioterapia continúa siendo una herramienta importante, especialmente el alivio de la sintomatología secundaria a fracturas patológicas. Se estima que hasta el 38% de los pacientes con MM recibirán radioterapia paliativa en el curso de su enfermedad. Las indicaciones para esta opción terapéutica son: prevención y tratamiento de fractura no corregible por medios quirúrgicos, dolor intenso, compresión medular, afección a órgano vital, enfermedad extramedular y paliación de otra sintomatología relacionada con fracturas. Se utilizan dosis de entre 10 y 30 Gy fraccionadas, con tasas de respuesta de hasta el 76%, por lo menos en lo que respecta a la paliación del dolor6.
Independientemente de la opción disponible en nuestro centro, es importante tener en cuenta el manejo de esta complicación debido a su gran impacto en la calidad de vida y la experiencia global que el paciente experimenta con el tratamiento que proporcionamos para su patología neoplásica.
MANEJO DE DOLOR
El dolor oncológico puede deberse a progresión tumoral (obstrucción linfática y vascular, distensión, edema, inflamación y necrosis). La infiltración ósea tumoral es la causa más frecuente de dolor en el paciente hematológico. Los mecanismos causantes de dolor óseo se explican por la producción de prostaglandina E 2 (PGE2), que sensibiliza los nociceptores. La destrucción ósea lleva a fracturas patológicas e invasión perineural. Dentro de las enfermedades hematológicas, el MM es el que tiene mayor incidencia de dolor óseo, afectando hasta al 75% de los pacientes2.
Para reducir el dolor óseo la manera más eficaz es controlar la enfermedad de base, pero en ocasiones es necesario recurrir al uso de analgésicos convencionales como los antiinflamatorios no esteroideos (AINE), opioides y fármacos adyuvantes (paracetamol, anestésicos locales, esteroides, antidepresivos, neuromoduladores o agonistas alfa 2, entre otros). El tratamiento del dolor se puede sistematizar de acuerdo con lo propuesto en la escalera analgésica de la Organización Mundial de la Salud (OMS)3.
Para poder entender el tratamiento del dolor, es importante conocer su clasificación. De acuerdo con su fisiopatología, se clasifica en nociceptivo (somático) y neuropático. El dolor nociceptivo es aquel dolor causado por la estimulación de fibras Ad y C por un estímulo nocivo sobre los tejidos corporales, es una respuesta fisiológica a una agresión. El dolor somático se origina por lesiones en piel, músculo, hueso y cápsulas articulares, y suele ser bien localizado. El dolor óseo se engloba en esta categoría. El dolor neuropático se origina como consecuencia directa de una lesión o enfermedad que afecta el sistema nervioso central o periférico4.
Con lo anterior, es importante también mencionar el esquema de analgesia propuesto por la OMS (Fig. 1).

Figura 1 Esquema de analgesia de la OMSAINE: antiinflamatorio no esteroideo; EVA: escala visual analógica.
Para poder evaluar la intensidad del dolor contamos con diferentes escalas, sin embargo, la más sencilla de aplicar es la escala verbal analógica (EVA) o cualquier otra escala analógica unidimensional para la evaluación de dolor.
El uso de AINE se limita a pacientes cuya tasa de filtrado glomerular (TFG) sea mayor a 60 ml/min y la recomendación es utilizarlos en un periodo máximo de siete días consecutivos. Por esto es importante mencionar que el manejo farmacológico del dolor óseo en el paciente con MM tiene su base en los opioides.
Podemos clasificar este grupo de medicamentos por su potencia como se muestra en la tabla 2.
Tabla 2 Clasificación de Analgésicos.
| Débiles | Potentes |
|---|---|
| Nalbufina | Sufentanilo |
| Tapentadol | Fentanilo |
| Tramadol | Buprenorfina |
| Codeína | Hidromorfona |
| Dextropropoxifeno | Oxicodona |
La morfina se considera el estándar de oro de los opioides y con base en este se determina la potencia7.
Se sugiere iniciar el manejo del dolor con opioides débiles y ajustar la dosis; sin embargo, si el dolor es muy intenso podría iniciarse con opioide potente para control de dolor y, de igual manera, ajustar la dosis.
Las dosis iniciales recomendadas para manejo de dolor son las siguientes:
‒ Nalbufina: 10-20 mg cada 3-6 horas. Dosis máxima 160 mg/24 h8.
‒ Tapentadol: 50-100 mg cada 6 horas. Dosis máxima 500-600 mg/24 h9.
‒ Tramadol: 1-3 mg/kg. Dosis inicial 25 mg. Dosis de mantenimiento 50-100 mg cada 6 horas. Dosis máxima 600 mg/24 h. Dosis ajustada a función renal: 200 mg/24 h máximo8.
‒ Morfina: 10-15 mg/12 h (pacientes vírgenes a opioides) y 20-30 mg/12 h (pacientes previamente expuestos a opioides). Dosis inicial 5 mg/4 h. En pacientes con enfermedad renal leve o moderada es necesario hacer un ajuste de dosis del 25 al 50% de la dosis basal10.
‒ Oxicodona: 5 mg/4 h. (1 mg de oxicodona = 2 mg de morfina). Su uso es más seguro en pacientes con insuficiencia renal11.
‒ Buprenorfina: 0.3-0.6 mg/6-8 h. Dosis máxima 140 µg/h (3,360 µg/24 h).
‒ Fentanilo: 0.04 µg/kg como dosis inicial y 0.5-1 µg/kg/h para mantenimiento9.
Es importante señalar la acción dual de opioides como el tramadol y el tapentadol9.
En la tabla 3 se muestran los ajustes de dosis de los fármacos analgésicos ajustados a función renal para un uso seguro en el paciente con MM.
Tabla 3 Fármacos para el tratamiento del dolor y su utilización en la insuficiencia renal, valorada según el grado de insuficiencia renal por filtrado glomerular (FG) (ml/min/1.73 m2)
| Grupo y fármacos | FG (ml/min/1.73 m2) | Hemodiálisis | ||||
|---|---|---|---|---|---|---|
| Leve (> 60) | Moderada (30-60) | Grave (< 30) | ||||
| Fentanilo | Fentanilo Effentora© Actiq© Abstral© Pecfent© Instanyl© |
Seguro con reducción de dosis
Escasamente dializable Precaución |
||||
| Buprenorfina | Transtec© | |||||
| Tramadol | 10-30 ml/min: 200 mg/24 h > 10 ml/min: 50 mg/24 h |
Elimina cantidades mínimas de tramadol | ||||
| Tramadol de liberación prolongada | Elimina cantidades mínimas de tramadol | |||||
| Morfina | Un 25% en FG 30-59 ml/min | 50% FG 15-29 ml/min y 75% FG > 15 ml/min | Precaución y ajuste de dosis
Dializable |
|||
| Morfina de liberación prolongada | ||||||
| Oxicodona Oxicodona/naloxona |
Oxycontin© Targin© | Necesario ajustar dosis en IR
Iniciar con la mitad (50%) de la dosis habitual e ir
ajustando dosis En el caso de los pacientes con IR grave es particularmente necesaria una vigilancia médica estricta |
||||
| Jurnista© | Necesario ajustar dosis en IR moderada Iniciar con un tercio a la mitad de la dosis habitual e ir ajustando dosis | Se convierte a su metabolito menos activo, que además se elimina fácilmente con la hemodiálisis | ||||
| Tapentadol retard | Yantil©
Palexia© |
No se recomienda su uso en IR grave (FG < 30 ml/min) | ||||
| Codeína | Necesario ajustar dosis en IR moderada
Iniciar con un 75% de la dosis habitual |
No se recomienda su uso en IR grave (FG < 30 ml/min) | ||||
| Gabapentina | 50-79 ml/min: 600 mg/8 h | 30-49 ml/min: 300 mg/8 h | 15-29 ml/min: 300 mg/12 h
< 15 ml/min: 300 mg/24 h |
Con precaución | ||
| Pregabalina | > 60 ml/min: 300 mg/12 h | > 30-59 ml/min: 150 mg/12 h | 15-30 ml/min: 150 mg/24 h
< 15 ml/min: 75 mg/24 h |
Con precaución | ||
| Paracetamol | < 10 ml/min: espaciar dosis cada 8 h | |||||
| AINE | Se debe evitar el uso innecesario en IR | |||||
IR: insuficiencia renal; AINE: antiinflamatorio no esteroideo.
Adaptada de Terpos, et al., 20135.
Una vez iniciado el manejo farmacológico del dolor, o en caso de no tener una respuesta favorable o no contar con los recursos farmacológicos apropiados, se sugiere referir al paciente a una clínica de cuidados paliativos para manejo transdisciplinario de síntomas.
Dolor neuropático: origen, manifestaciones clínicas y tratamiento
El MM es una enfermedad con alta carga sintomatológica y puede condicionar a que el paciente presente neuropatía y dolor neuropático, el cual tiene características diferentes en la percepción y referencia por parte del paciente con respecto al dolor óseo.
Las características del dolor neuropático incluyen: parestesias, disestesias, dolor quemante, alteración en la sensibilidad al tacto, contracción muscular y alteraciones en la piel (como hiperestesia, hipoestesia y parestesia). Localización: en manos y pies. Se acompaña de alteraciones del sistema nervioso autónomo, atrofia muscular, pérdida de la fuerza, atrofia de los músculos distales, hipotensión ortostática, bradicardia e hipotensión11.
Tratamiento del dolor neuropático en el paciente con mieloma múltiple
Existen muchas guías terapéuticas para el tratamiento del dolor neuropático. Los medicamentos que se prescriben comúnmente son: antidepresivos tricíclicos, inhibidores selectivos de la recaptación de la serotonina (ISRS), anticonvulsivantes y opioides11.
-
‒ Primera línea de tratamiento.
-
Antidepresivos tricíclicos:
Amitriptilina 25-150 mg/24 h.
Nortriptilina 25-150 mg/24 h.
-
Inhibidores selectivos de la recaptación de noradrenalina y serotonina:
Duloxetina 30 mg/24 h-60 mg/12 h.
Venlafaxina 37.5-225 mg/24 h.
Bloqueadores de los canales de calcio a2-d.
Pregabalina en dosis bajas, con la finalidad de reducir los efectos secundarios como somnolencia, mareo y náuseas. En dosis iniciales de 25-75 mg cada 24 h y aumentando la dosis de 150 mg, 300 mg, hasta una dosis máxima de 600 mg al día divido en dos dosis.
Gabapentina: la dosis inicial es de 300 mg/d incrementados en 300 mg cada 3-7 días, hasta dosis de 900-3,600 mg/d en intervalos de 8 h. Los efectos secundarios más frecuentes, moderados y normalmente bien tolerados, son somnolencia, vértigos, cefalea y diarrea.
Parche de lidocaína al 5% (700 mg): colocar el parche sobre la zona afectada durante 12 h, la dosis máxima es de tres parches.
-
-
‒ Segunda línea: opioides.
Tramadol: debido a que tiene dos mecanismos de acción, la unión a receptores opioides y la inhibición de la recaptación de noradrenalina y serotonina, siendo este segundo efecto el más potente. Dosis de 50-600 mg/24 h dividido en tres dosis.
Tapentadol: 50 mg dos veces al día, titulando hasta una dosis máxima de 250 mg dos veces al día, con una dosis máxima de 700 mg día. La dosis habitual es de 50 mg cada 6-8 h, con una dosis máxima de 400 mg/d.
Opioide potente: utilizar la oxicodona, que tiene buen resultado en el tratamiento del dolor neuropático. Iniciar con dosis de 5-10 mg/12 h.
Sulfato de morfina: iniciar con dosis bajas, de 15-30 mg en 24 h12.
-
‒ Medidas de soporte:
Generalmente estos pacientes presentan dificultad para la ejecución de las actividades de la vida diaria, por lo que se benefician con ejercicios de rehabilitación y fisioterapia.
Como apoyo para la prevención de la constipación inducida por opioides: ingesta de fibra y líquidos, así como de laxantes, hidratación y ajuste de fármacos1.
Debido a la gran variedad de factores causales y contribuyentes en la patogenia de la neuropatía, no existe un tratamiento único satisfactorio para esta entidad. El alivio de esta manifestación puede ser uno de los problemas más difíciles que afrontan el médico y el paciente. Las diferencias genéticas en las vías del dolor parecen explicar por qué distintos tipos de medicamentos que inhiben de manera selectiva vías específicas pueden ser efectivos en algunos pacientes, pero en otros no.
Neurotoxicidad inducida por quimioterapia
La neurotoxicidad es común en pacientes oncológicos, ya sea por la enfermedad o por los tratamientos para esta. Es frecuente que involucre diferentes estructuras del sistema nervioso, como los nervios sensoriales periféricos, que son los más afectados y producen dolor, cuya intensidad depende del tipo y acumulación del fármaco, además de ser dependientes de la dosis. Es más frecuente en pacientes con neuropatía preexistente. Los síntomas dependerán de las fibras afectadas, pero lo más habitual es que se presente con alteraciones en la percepción de la temperatura y dolor; la afección suele ser simétrica, similar a la distribución de la neuropatía diabética, en guante y calcetín, siendo los pies los principalmente afectados13.
Además del tratamiento previamente mencionado, el manejo de la neuropatía inducida por quimioterapia se ve beneficiado del uso de vitamina E a dosis de 200-400 mg cada 12 horas14.
Manejo de otros síntomas de mieloma múltiple
Desafortunadamente, no son los únicos síntomas que afectan a los pacientes con MM. Dentro de las etapas tempranas de la enfermedad, los síntomas que más afectan la calidad de vida son el dolor y los síntomas psicoafectivos, como la ansiedad y depresión. En etapas más avanzadas se ha reportado fatiga extrema y mucositis, en el caso de manejo con melfalán y ciclofosfamida, principalmente14.
Mucositis
La mucositis se define como la reacción secundaria a la quimioterapia o radioterapia y se caracteriza por la presencia de áreas eritematosas, lesiones ulcerativas en la mucosa oral que causan dolor y limitación a la alimentación. Este es uno de los efectos secundarios más comunes dentro del tratamiento oncológico, alrededor del 40% de los pacientes desarrollarán mucositis, dependiendo del agente quimioterápico empleado15
Para la clasificación de la mucositis se puede utilizar la escala que se muestra en la tabla 4.
Tabla 4 Mucositis según la Organización Mundial de la Salud (OMS)
| Grado | Mucositis según la OMS |
|---|---|
| 0 | Sin lesión |
| 1 | Mucosa rosada, no dolorosa, con abundante saliva |
| 2 | Eritema, úlceras poco extensas, se mantiene la deglución |
| 3 | Úlceras extensas, encías edematosas, saliva espesa, se mantiene la deglución de líquidos, dolor y dificultad para hablar |
| 4 | Úlceras muy extensas, encías sangrantes, infecciones, no hay producción de saliva, lo que imposibilita la deglución, dolor muy intenso |
MEDIDAS NO FARMACOLÓGICAS
Promover el aseo oral mediante el uso de agentes no abrasivos: aloe vera, miel de abeja, bicarbonato y manzanilla; estos agentes promueven la cicatrización, retardan la evolución de la lesión y evitan la progresión de estado avanzados, por su propiedades favorecedoras de la producción de saliva, antiinflamatorias, antioxidantes y antibióticas15.
TERAPIA FARMACOLÓGICA
Fármacos protectores de mucosa, bencidamina, fosfato de calcio, clorhexidina, crioterapia, factores de crecimiento, terapia láser, higiene oral1.
-
‒ Colutorios: fármacos citoprotectores combinados con anestésicos locales y antihistamínicos; forman una placa protectora sobre la mucosa, incrementado la secreción de PGE2; favorecen la reparación tisular con propiedades antiinflamatorias y analgésicas15.
Se recomienda utilizar la siguiente fórmula magistral:
5 ml lidocaína 1%.
30 ml de hidróxido de aluminio y magnesio.
30 ml de jarabe de difenhidramina o de clorfenamina15.
‒ Bencidamina: disminuye la severidad de la lesión y la sintomatología dolorosa.
‒ Enjuagues de fosfato de calcio o clorhexidina: disminuyen las lesiones y el dolor, por lo cual mejora la ingesta de alimentos.
‒ Crioterapia: provoca vasoconstricción, reduciendo el tamaño de la lesión y disminuyendo la sintomatología dolorosa.
Fatiga
De igual manera, para entender el tratamiento de la fatiga es necesario conocer su causa, que en el caso del MM es multifactorial y generalmente se asocia con insomnio. El estar fatigado y no poder dormir tiene un alto impacto en los índices de calidad de vida y el estado de ánimo de los pacientes.
La fatiga se define como como un síntoma que puede ser derivado de la enfermedad o de sus tratamientos, impactando de manera negativa en la calidad de vida de los pacientes. Las manifestaciones clínicas de la fatiga incluyen bajos niveles de energía, sentirse débil o letárgico, concentración reducida y afectación de la memoria, cambios abruptos del estado de ánimo, irritabilidad y labilidad emocional, mareo, desorientación, disnea, insomnio, alteración en la libido y pérdida de apetito.
El origen de la fatiga en el caso de MM es multifactorial y dentro de sus causas se encuentra la anemia, alteraciones en la insuficiencia renal, dolor, infecciones asociadas, poca ingesta de alimentos, alteraciones metabólicas (por liberación de citocinas por la enfermedad), alteraciones en el estado de ánimo y los tratamientos hematológicos. Es importante tratar cada una de las causas para controlar este síntoma y así propiciar una mejor calidad de vida en este grupo de pacientes16.
Dentro del tratamiento para la fatiga tenemos medidas farmacológicas y no farmacológicas. Entre las no farmacológicas tenemos el reposo y descanso de los pacientes durante el día, con horarios limitados para que no interfieran con el ciclo circadiano; las transfusiones sanguíneas para corrección de anemia, tema que será abordado en el apartado de profilaxis y manejo de citopenias, y mantener una hidratación y alimentación apropiada, en ocasiones requiriendo suplementos alimenticios. Dentro de las medidas farmacológicas tenemos el uso de opioides y fármacos adyuvantes para control de dolor (descrito previamente en el apartado de dolor) y antibióticos en caso de presentar infecciones (los esquemas deberán usarse de acuerdo con la sensibilidad y el microorganismo que las ocasione)17.
Insomnio
El insomnio es un síntoma presente hasta en un tercio de los casos de los pacientes hematooncológicos en algún momento de la enfermedad. Se relaciona estrechamente con la existencia de depresión, ansiedad o síntomas físicos no controlados. También puede aparecer por sí mismo sin que sea expresión de un trastorno psiquiátrico, como efecto secundario de otros tratamientos. Su tratamiento se basa en la corrección de la causa cuando sea posible, en una adecuada higiene del sueño (colocar relojes, calendarios, mantener iluminado el cuarto con luz natural durante el día y completamente oscuro durante la noche, establecer horarios y rutinas para dormir) y en el uso de hipnóticos benzodiacepínicos, no benzodiacepínicos o neurolépticos si concurre una situación de demencia o delirium. En ocasiones pueden también utilizarse inductores del sueño como la melatonina18.
Síntomas psicoafectivos
En el manejo de los síntomas psicoafectivos se deben considerar dos aspectos importantes para manejarlos de manera apropiada: el farmacológico (que será abordado en este apartado) y el no farmacológico (que será abordado en el apartado de abordajes psicoemocionales en pacientes y cuidadores primarios informales).
DEPRESIÓN
De acuerdo con la OMS, es un trastorno del estado de ánimo que se caracteriza por la presencia de tristeza, pérdida de interés o placer, sentimientos de culpa o falta de autoestima, trastornos del sueño o del apetito, sensación de cansancio y falta de concentración18.
Existen condiciones clínicas que suelen asociarse a la presencia de sintomatología depresiva en los pacientes con MM, como las alteraciones del calcio, del sodio y del potasio, anemia, déficit de vitamina B12, enfermedades existentes y el uso de fármacos: esteroides, interferón, algunos antibióticos, agentes quimioterápicos (vincristina, vinblastina, asparaginasa, metotrexato intratecal, interleucina, anfotericina B, tamoxifeno).
Los antidepresivos son los fármacos indicados en el manejo del episodio depresivo y sus manifestaciones clínicas. Los grupos de antidepresivos en los que hay mayor evidencia científica son los ISRS y los antidepresivos tricíclicos18.
Profilaxis y manejo de citopenias
Neutropenia
La neutropenia es un efecto adverso de muchos fármacos utilizados en la oncohematología, así como una manifestación inicial de varias enfermedades. El desarrollo de guías para el manejo de esta complicación está orientado a su profilaxis y manejo en tumores sólidos y enfermedades que requieren quimioterapia intensiva, como las leucemias agudas; no existen guías específicas de neutropenia en MM, por lo tanto, adaptaremos aquello que nos es útil de las guías existentes.
El paso inicial para determinar si un paciente requiere o no profilaxis con factores estimulantes de colonias de granulocitos (G-CSF) en la determinación del riesgo de neutropenia. Esta determinación se toma con base en el manejo utilizado y las características del paciente. Consideraremos factores de alto riesgo para neutropenia a las quimioterapias aplasiantes (antraciclinas, antimetabolitos; el único esquema de uso de MM considerado de alto riesgo es DT-PACE), uso de anticuerpos monoclonales, edad mayor de 60 años, pobre status físico, infecciones activas al momento del uso de la quimioterapia y los que presentan insuficiencia hepática o renal. Este grupo de pacientes se considera que tiene un 20% de riesgo de desarrollar neutropenia febril y requiere de profilaxis con factores estimulantes. Los pacientes que no entren en esta categoría se considerarán de riesgo intermedio, con un 10% de riesgo de desarrollar neutropenia febril, y en ellos quedará, a decisión del terapéutico, el uso de profilaxis con factores estimulantes.
La elección del factor a utilizar (G-CSF vs. G-CSF pegilado) recaerá en la disponibilidad del tratamiento. El G-CSF es uno de los más ampliamente utilizados, la dosis recomendada es 5 µg/kg de peso, 3 a 4 días posteriores a la quimioterapia aplasiante o hasta que se resuelva la neutropenia. La dosis recomendada de factores pegilados es de 6 mg, dosis única un día después de terminada la quimioterapia. En caso de presentarse neutropenia febril se deberá utilizar el mismo factor estimulante que se utilizó para la profilaxis, tomando en cuenta que no existe evidencia certera de que esta intervención tenga algún impacto en la mortalidad, más si en la duración de la neutropenia las dosis a utilizar son las mismas que para profilaxis, pero acompañadas del esquema antibiótico adecuado a cada caso.19
Anemia
La anemia es, per se, una manifestación del MM y de algunas de las gammapatías estudiadas, su vigilancia es uno de los puntos clave para el seguimiento de estos pacientes, por lo tanto, previo a decidir el uso de algún factor estimulante se debe descartar que la citopenia mencionada no sea una manifestación de progresión de la enfermedad.
Es sabido que la eritropoyetina y sus análogos aumentan el riesgo de trombosis, riesgo de por sí ya elevado por la biología y tratamiento de la enfermedad, por lo que su uso deberá discutirse y sopesarse con este riesgo en casos en los que se haya demostrado cabalmente que la anemia es secundaria a alguno de los esquemas terapéuticos utilizados.
En este caso se debe de considerar transfusión de concentrados eritrocitarios (CE) para el manejo del síndrome anémico, de acuerdo con la guía de uso clínico de la sangre. De esta manera se debe transfundir CE a todo paciente con hemoglobina inferior a 10 g y que tenga datos clínicos de hipoxia tisular (palidez tegumentaria, mareo, disnea de medianos a pequeños esfuerzos). La dosis recomendada dependerá de cada paciente y de las comorbilidades que este tenga, y se deberá poner especial atención en aquellos pacientes sometidos a trasplante de células progenitoras hematopoyéticas (TCPH) o postrasplantados, en los cuales la indicación es la transfusión de CE radiados.
En caso de que el paciente sea altamente dependiente de transfusión de CE, se podrá valorar el uso de análogos de la eritropoyetina como medida de tratamiento, la cual ha demostrado disminuir la dependencia transfusional, pero no impactar positivamente en el pronóstico de la enfermedad19.
Trombocitopenia
La trombocitopenia puede ser también un dato inicial de la enfermedad, con menor frecuencia que la anemia. La administración de concentrados plaquetarios o aféresis plaquetarias profilácticas está indicada en los siguientes casos: pacientes estables clínicamente con conteos plaquetarios menores a 10,000 plaquetas secundario a quimioterapia, TCPH o actividad de la enfermedad; pacientes con menos de 20,000 plaquetas con infecciones activas, alteraciones en la coagulación, ya sea secundario al tratamiento o a la enfermedad, y con antecedente de sangrado masivo.
La dosis para administrar el concentrado plaquetario será de 5 a 8 CP cada 24 h o una aféresis plaquetaria cada 24 h hasta la resolución del factor de riesgo para sangrado o la normalización de la cifra plaquetaria. En el caso de pacientes sometidos a TCPH se deberán administrar productos plaquetarios leucorreducidos19.
Abordajes psicoemocionales en pacientes y cuidadores primarios e informales
Desde el modelo biopsicosocial, se asume que una enfermedad comprende no solo alteraciones a un nivel orgánico, sino que además existe un proceso de cambio y adaptación a nivel psicológico, familiar, social, laboral y espiritual que considera la historia del desarrollo del paciente, así como su contexto social, histórico, político y cultural. Es así que el abordaje del MM implica la integración de disciplinas complementarias a la medicina, como lo es la psicología, que en el ámbito de la salud lleva varias décadas implementando intervenciones en los tres niveles de atención a la salud, que oscilan desde la prevención y promoción de la salud hasta la atención en cuidados paliativos y el bien morir, poniendo énfasis especial en el comportamiento, ya que este desempeña un papel fundamental en el proceso de salud-enfermedad20.
Primero es necesario considerar que informar sobre el diagnóstico de MM a un paciente supone dar una mala noticia que provoca una alteración del significado y las expectativas del presente y el futuro de la vida del paciente21. Este diagnóstico supone que la enfermedad adquiere la cualidad de estresor, puesto que el paciente la evalúa como una amenaza a su organismo y, de esta forma, automáticamente se generan patrones de respuestas que están en función de las diferencias individuales que cada paciente posee, como los estilos de afrontamiento y la percepción de recursos disponibles para afrontar esta amenaza22,23.
Estas respuestas al estrés se pueden observar en cambios a nivel cognitivo, emocional, fisiológico y comportamental, y en el contexto de un diagnóstico como el MM existe una serie de respuestas que pueden ser esperadas debido a la relevancia de esta situación en la vida del paciente, como llanto, enojo, tristeza, ansiedad, miedo, pensamientos negativos, aislamiento social, irritabilidad, exacerbación del dolor y aumento de disnea, entre otras.
De esta forma, la monitorización constante del paciente y su familia por parte del equipo de salud es una tarea de suma importancia, debido a que el MM adquiere el carácter de estresor crónico, lo que puede generar respuestas desadaptativas y evolucionar a estados patológicos. Es así que el apoyo que el paciente reciba, ya sea de su familia, amigos y equipo médico, adquiere un rol fundamental en el curso de la enfermedad, pues permite que el paciente desarrolle patrones adaptativos de respuesta y disponga de los recursos necesarios para afrontar de forma efectiva las demandas de la enfermedad, y así, mejorar la adaptación a esta24.
A pesar de que los avances terapéuticos en el tratamiento del MM han logrado un aumento de la supervivencia libre de progresión y las tasas de supervivencia global, los efectos propios de la enfermedad y la toxicidad producida por el tratamiento, también existe una mayor intensidad de síntomas y efectos negativos propios de la historia natural de la enfermedad, lo que se traduce en una menor calidad de vida del paciente. Por lo cual, uno de los mayores retos en la actualidad es lograr un equilibrio entre la carga de los síntomas propios de la enfermedad y derivados de los tratamientos con el mantenimiento de la funcionalidad y la calidad de vida del paciente25.
En consideración a lo anterior, actualmente existe el reconocimiento de necesidades de atención psicológica en los pacientes con cánceres hematológicos como el MM, debido a que por sí mismo representa un factor de riesgo para el desarrollo de problemas de salud mental26. Las necesidades psicológicas no tienen un curso especifico de comportamiento, por el contrario, puede presentarse desde el diagnóstico, durante el tratamiento y en los últimos días de vida, por lo que la evaluación que el personal de salud realice representa un factor clave en el reconocimiento y atención dichas necesidades27.
Desde el ámbito de la medicina paliativa, estas necesidades psicosociales adquieren especial relevancia en la atención a los pacientes y también a sus familiares, por lo cual las intervenciones psicológicas están orientadas a28:
‒ Manejo de síntomas como dolor, náuseas, vómito, insomnio y disnea.
‒ Psicoeducación sobre la enfermedad, síntomas, tratamientos y efectos adversos.
‒ Manejo del estado emocional en problemas como depresión o ansiedad.
‒ Desarrollo de conductas protectoras y de autocuidado como la adherencia al tratamiento y la actividad física.
‒ Atención a problemas sexuales.
‒ Fortalecimiento de la autoestima, la espiritualidad y el sentido de vida.
‒ Manejo del duelo y duelo anticipado.
‒ Mejora en relaciones interpersonales y optimización de recursos familiares.
‒ Mantenimiento de los valores de vida, la esperanza y la dignidad.
De esta forma, uno de los retos actuales para la psicología como disciplina ha sido adaptar las intervenciones para que se ajusten a las condiciones de fragilidad con las cuales se trabaja desde la medicina paliativa29, como se observa en la Figura 2 en cada etapa de la evolución de la enfermedad hay intervenciones psicológicas especificas.

Figura 2 Objetivos de las intervenciones psicológicas de acuerdo con las diferentes fases de la enfermedad.
La multifactorialidad del MM cobra especial relevancia en su atención, ya que se debe abordar desde diversos aspectos, comenzando por la parte física y hasta el área psicoemocional, para brindar al paciente calidad de atención durante su enfermedad.
Es importante tomar en cuenta la estructura psicológica y de personalidad del paciente, así como la existencia de psicopatologías previas; la experiencia directa con la enfermedad puede influir en la respuesta emocional del paciente.
Considerando lo anterior, resulta necesario contar con un protocolo de atención psicológica que permita intervenir al paciente desde una fase inicial y poder acompañarlo en el proceso de su enfermedad.
Factores asociados a la alteración emocional de pacientes con mieloma múltiple
‒ Antecedentes de trastornos psiquiátricos (depresión, ansiedad y consumo de sustancias, entre otros).
‒ Ansiedad persistente desde el inicio de la enfermedad y en el transcurso de esta, con recursos de afrontamiento limitados.
‒ Redes de apoyo limitadas, aumentando la percepción de amenaza y respuestas fisiológicas ante el estrés.
‒ Mal apego a tratamientos médicos, que derivan en descontrol de síntomas físicos y psicológicos.
‒ Pobre comunicación con el equipo médico, imposibilitando la alianza terapéutica con el equipo de salud.
Fase de diagnóstico
En la fase de diagnóstico la intervención del psicólogo va encaminada a detectar las necesidades y déficits del paciente y su familia, así como dotar de habilidades de comunicación al paciente y su familia para establecer metas en común y objetivos de tratamiento con el equipo médico, además de mantener un nivel de información adecuado a las necesidades personales del paciente30.
Además, el psicólogo interviene brindando herramientas funcionales al paciente para proporcionar la percepción de control sobre la enfermedad, como promover la integración de los estilos de afrontamiento del paciente que le permitan mantener su funcionalidad cotidiana e iniciar el entrenamiento para el manejo del estrés, la ansiedad y otros síntomas psicológicos que pueden presentarse debido a la notificación del diagnóstico de la enfermedad. Asimismo, es importante apoyar a los cuidadores primarios informales y familiares en la solución de problemas relacionados con la nueva situación de salud del paciente.
Fase de tratamiento
En esta etapa es importante apoyar al paciente en el conocimiento de la enfermedad, así como en los síntomas asociados a esta y los tratamientos propuestos por el equipo médico por medio de sesiones de psicoeducación, donde se brinde información relevante, clara y efectiva. Asimismo, se debe proporcionar un espacio abierto para la resolución de dudas con el equipo médico, logrando una alianza terapéutica con el equipo de salud que derivará en el apego a tratamientos31.
Aunado a lo anterior, abordar desde la psicología los síntomas físicos asociados a la enfermedad (náusea anticipatoria, dolor, etc.) y las reacciones emocionales esperadas (ansiedad, depresión, pérdida del sentido de vida, etc.) mediante intervenciones basadas en evidencia, como terapia cognitiva conductual, técnicas de relajación, técnicas de reestructuración cognitiva, mindfulness o terapia centrada en el significado facilitan la adaptación a la enfermedad durante el proceso de tratamiento32.
Es igualmente relevante fomentar la creación de redes de apoyo social efectivas en esta fase, ya que serán de suma importancia para incidir directamente en la adherencia a tratamientos y en la prevención del síndrome de cuidador primario33.
Fase de intervalo libre de enfermedad
La intervención del psicólogo en esta etapa cobra relevancia, ya que el paciente puede presentar alteraciones emocionales relacionadas con experiencias previas con la enfermedad y el temor a que se presenten de nuevo. Esto también puede reflejarse en una constante atención a las reacciones de su cuerpo, lo que puede crear actitudes hipocondriacas, derivando en problemas como ansiedad y depresión.
Fase de trasplante
La literatura refiere que antes del ingreso a un trasplante, el 40% de los pacientes presentan ansiedad, que puede desaparecer durante el seguimiento, sin embargo, persiste la depresión como síntoma predominante asociado a las estancias prolongadas en el hospital, así como los síntomas físicos que se pueden presentar. De igual forma, el aislamiento forma parte importante de los factores que fomentan síntomas depresivos.
Es importante tomar en cuenta que el entrar a una unidad de trasplante puede derivar en diversas emociones, como angustia, miedo, confusión, esperanza e incertidumbre, y existirá un ajuste psicológico importante. Uno de los momentos reportados como más angustiantes para pacientes y familia que son sometidos a un trasplante es la firma del consentimiento informado, donde se describen todos los detalles del procedimiento y las posibles consecuencias sobre el paciente34.
Aquí, el rol del psicólogo es explorar los conflictos que presenta en cada fase del trasplante para realizar una intervención detallada dirigida al paciente y su familia, como la hospitalización de aislamiento, que puede prolongarse hasta varios meses. La intervención psicológica estará dirigida a la adaptación del paciente a estas condiciones de su tratamiento, además de desarrollar y potenciar recursos y estrategias que favorezcan la adherencia a este y realizar intervenciones desde el enfoque cognitivo conductual.
Fase de recidiva
El impacto emocional puede ser mucho más fuerte que el diagnóstico y pueden presentarse diversos síntomas de difícil control para el equipo de psicología. Esta etapa está encaminada a brindar contención y validación emocional, contribuyendo a prevenir y detectar problemas de salud mental, brindar estrategias de apoyo para la adaptación a este estado de enfermedad, apoyar en la relación médico-paciente para crear alianzas fortalecidas, así como abrir canales de comunicación para la expresión y resolución de dudas respecto a esta nueva etapa.
Fase paliativa
Esta etapa toma especial relevancia para el acompañamiento del paciente y familia, ante la pérdida irremediable de salud y la limitación que esto conlleva. Se debe acompañar a la familia y apoyar en estas nuevas necesidades, así como la movilización de recursos para el manejo de emociones adversas como ira, tristeza, rumiaciones y repercusiones en la autosuficiencia, entre otras, por lo cual es importante poder detectar las necesidades específicas del paciente y priorizar aquellas que son de relevancia.
En esta fase final de la enfermedad, los síntomas físicos y emocionales, cognición, necesidades espirituales y psicológicas toman relevancia, por lo cual se debe poner atención en el manejo de emociones, la comunicación entre médico y familia y médico y paciente, la toma de decisiones y las atenciones del cuidador primario.
En este sentido, es importante prevenir la sobrecarga del cuidador, fomentando el autocuidado y los hábitos saludables, así como fomentar la planificación y organización de la familia en los cuidados del enfermo.
Asimismo, una de las intervenciones del psicólogo va enfocada a acompañar a la familia en la elaboración del duelo, por medio de diversas intervenciones clínicas (reuniones familiares, psicoeducación, terapia ajustada a necesidades del familiar) que pueden fomentar la comunicación activa y resolución de conflictos con el paciente.
Espiritualidad
La espiritualidad se reconoce generalmente como abarcadora de aspectos experienciales, ya sea relacionados con la participación en prácticas religiosas o bien con el reconocimiento de una sensación general de paz y conectividad. La espiritualidad es un recurso para trascender el sufrimiento que acompaña la pérdida y para promover la aceptación en el proceso de morir.
El cuidado espiritual se basa en el modelo de cuidado bio-psico-social-espiritual, y en una perspectiva integral de la persona, reconociendo y potenciando los recursos de los pacientes y facilitando la sanación al trascender el sufrimiento Los pacientes aceptan que los profesionales se interesen por sus necesidades espirituales y desean ser apoyados en el proceso de morir.
Es importante validar las inquietudes espirituales del paciente, esto puede ser de forma verbal o bien por medio de una evaluación escrita. Existen diversos instrumentos que evalúan sistemas de creencias espirituales, integración de una comunidad espiritual, implicaciones en la atención médica y planificación de eventos al final de la vida, entre otros. El abordar la parte espiritual tiene implicaciones en la mejora de la calidad de vida y la mayor probabilidad de recibir atención al final de la vida.35
Módulo 9: Tromboprofilaxis y hemostasia en el mieloma múltiple
Es bien conocido que el cáncer es un factor de riesgo para la presentación de trombosis, con un aumento en el riesgo de cuatro a siete veces en comparación con la población general. El mieloma múltiple (MM) aumenta nueve veces el riesgo de trombosis al tratarse de una enfermedad maligna, debido a fenómenos propios de la fisiopatología de la enfermedad, tratamiento y factores propios del paciente, estimándose la incidencia de trombosis en el mieloma en 8 a 22 por cada 1,000 personas/año1.
El riesgo de trombosis venosa profunda (TVP) es mayor en los primeros 3-6 ciclos de quimioterapia en el paciente de reciente diagnóstico, comparado con la trombosis arterial, que aumenta después del año de diagnóstico2.
La fisiopatología de la trombosis en el MM depende de factores inherentes a la enfermedad, al paciente y al tratamiento utilizado.
Los factores relacionados con la enfermedad se dividen en:
‒ Componente monoclonal: incluyen producción de factores paraproteínicos que causan una viscosidad excesiva de la sangre, lo que conduce a una alteración de la estructura de la fibrina y al retraso de la fibrinólisis, así como a la producción de autoanticuerpos procoagulantes del tipo del anticoagulante lúpico.
‒ Hipercoagulabilidad: aumento del factor VIII, FVW (factor de von Willebrand) y fibrinógeno, disminución de las proteínas C y S, e incremento en el inhibidor del activador del plasminógeno-1 (PAI-1).
Las características más significativas del paciente que se consideran de riesgo son: la edad, mayor a 60 años; el sobrepeso; las comorbilidades cardiacas, renales, pulmonares y metabólicas; el antecedente de cirugía menor a seis semanas previas al diagnóstico; infección; accesos venosos centrales y coagulopatías1 Todo lo anterior se resume en la tabla 1.
Tabla 1 Factores de riesgo de trombosis venosa profunda (TVP) en paciente con mieloma múltiple
| Relacionados con el paciente | Relacionados con la enfermedad | Relacionados con el tratamiento |
|---|---|---|
| Edad | Relacionado con el componente monoclonal | IMiD |
| IMC > 30 | Hiperviscosidad | Dosis altas de dexametasona |
| Antecedentes familiares de TVP | Alteraciones de la malla de fibrina | Poliquimioterapia |
| Inmovilización | Anticoagulante lúpico | Doxorubicina |
| Comorbilidades | Inhibición de antitrombina, proteínas C y S | Catéter central |
| Cirugía reciente | Relacionado con el componente de la hipercoagulabilidad | |
| Catéter central | Incremento de FT, fibrinógeno, FVW y FVIII | |
| Infección aguda | Disminución de proteínas C y S | |
| Hospitalización | Incremento de PAI 1 |
IMiD: inmunomoduladores; IMC: índice de masa corporal; FT: factor tisular; FVW: factor de von Willebrand; FVIII: factor VIII;PAI 1: inhibidor del activador del plasminógeno-1.
El tratamiento es el tercer elemento que entra en juego al momento de definir el riesgo de trombosis en los pacientes con mieloma. Es bien sabido que el uso de quimioterapia y de esteroides a dosis altas, como por ejemplo dexametasona a dosis mayores a 480 mg por ciclo, deben ser tomados en cuenta al momento de decidir si dar o no tratamiento.
Especial atención merecen los inmunomoduladores (IMiD). Este grupo de medicamentos ha demostrado aumentar los eventos trombóticos en diversos grados, esto se debe a un aumento de la expresión del factor tisular, el factor de crecimiento vascular endotelial y la resistencia a la proteína C activada, entre otros, ejemplos claros son la talidomida, que se reporta con frecuencia de eventos trombóticos del 2 al 4% usada como monoterapia y hasta del 17% en combinación con quimioterapia y esteroides1.
En otros estudios el uso combinado de talidomida y dexametasona reportan hasta un 26% de trombosis, mientras que la combinación con antraciclina confiere un riesgo del 35%. Para el caso de la lenalidomida los reportes son similares.
Existen varias guías internacionales para el uso de tromboprofilaxis en enfermedades malignas, pero en la mayoría de estas el MM se considera de manera especial por su alta frecuencia de trombosis y el riesgo que implican sus características. No existen modelos predictores de riesgo específicos para estos pacientes, pero se han realizado esfuerzos tomando en consideración la cantidad de factores de riesgo con que se cuente al momento del diagnóstico, con el fin de orientar al clínico con respecto a qué medicamento usar para la profilaxis (Tabla 2).
Tabla 2 Factores de riesgo para trombosis
| Factor de riesgo |
|---|
| Factores individuales |
| - Obesidad (IMC > 30) |
| - Marcapasos |
| - Trombosis previa |
| - Enfermedades relacionadas |
| • DM |
| • HAS |
| • Nefropatía |
| • Infección |
| • Postración |
| - Cirugía |
| - Uso de EPO |
| - Coagulopatías |
| Factores relacionados con el mieloma |
| - Mieloma per se |
| - Hiperviscosidad |
| Factores relacionados con la terapia |
| - IMiD en combinación con: dosis alta de dexametasona, doxorubicina o QT combinada |
IMC: índice de masa corporal; DM: diabetes mellitus; HAS: hipertensión arterial sistémica; EPO: Eritropoyetina; IMiD: inmunomoduladores.
Adaptada de NCCN, 20206.
La decisión de utilizar aspirina vs. heparinas o cumarínicos se llevará a cabo con base en la cantidad de factores presentes al momento de la valoración y la ausencia de contraindicaciones para el uso de estos. La aspirina es el medicamento profiláctico utilizado con mayor frecuencia. El ácido acetilsalicílico (ASA) ha demostrado su eficacia en la prevención de eventos trombóticos en subanálisis de varios estudios de tratamiento; sin embargo, la información obtenida de estos estudios no es suficientemente robusta como para poder considerarlos fuentes de alta calidad para esta recomendación, pues en la mayoría de ellos se ha eliminado a pacientes con alto riesgo de trombosis3.
Las guías del International Myeloma Working Group (IMWG) y de la National Comprehensive Cancer Network (NCCN) admiten que el uso de ASA en monoterapia puede ser una maniobra útil en aquellos pacientes en riesgo bajo (un solo factor individual o relacionado con el mieloma) mas no en los que inician terapia con IMiD más quimioterapia3.
La heparina de bajo peso molecular y los cumarínicos han demostrado eficacia similar en la prevención de trombosis en un estudio comparativo entre estos medicamentos y el ASA4. En el caso de los cumarínicos, el international normalized ratio a seguir es entre 2 y 3; la ventaja de su administración oral y la existencia de antídoto, así como de estudios orientados específicamente a demostrar su eficacia en pacientes con MM los convierten en una excelente opción. Sin embargo, su seguimiento y manejo es complejo y puede tener repercusiones en la adherencia por parte del paciente. Las heparinas de bajo peso molecular disminuyen la frecuencia de trombos de igual manera que los cumarínicos, su gran desventaja es la vía de administración, y su ventaja es su relativa seguridad y poca interacción con otros medicamentos o alimentos; sin embargo, el hecho de ser inyectadas también impacta negativamente en la adherencia al tratamiento.
Desde hace ya varios años se ha desarrollado una amplia discusión con respecto al papel de los anticoagulantes directos (ACD) en el mieloma y el cáncer en general. Mientras que guías menos recientes, como la del IMWG, no recomendaban estos medicamentos debido a la falta de estudios apropiados para demostrar su seguridad, guías más recientes abren la posibilidad de su uso, especialmente en aquellos pacientes que tienen contraindicaciones para los anticoagulantes tradicionales, los que no desean recibir estos últimos o los que presentan resistencia a ellos. En el rubro en el que podemos encontrar más información al respecto es en el tratamiento de trombosis demostradas.
Los ACD han demostrado superioridad sobre la aspirina en el manejo posterior a la fase aguda de la trombosis, fase en la cual la indicación sigue siendo la heparina de bajo peso molecular (HBPM). Si bien la administración oral y el no requerir de monitoreo son condiciones atractivas para su uso, hay algunas objeciones que debemos considerar. Los pacientes con mieloma y cáncer en general tienen alteraciones en la absorción gastrointestinal de los medicamentos debido a la quimioterapia, las náuseas secundarias, el uso de múltiples fármacos y otras alteraciones; aunado esto al desconocimiento de las interacciones que pueden existir entre los ACD y los medicamentos utilizados para manejo del mieloma, su uso de manera sistemática puede ser dudoso5.
Hay un grupo especial en el cual los ACD pueden ser el tratamiento de primera elección (pacientes frágiles o distancias largas a sitios de muestras).
La NCCN, el IMWG y el grupo de la European Society of Medical Oncology han determinado algoritmos de manejo de acuerdo con el riesgo en que se encuentra el paciente, considerando de bajo riesgo a los que tengan un factor individual o relacionado con el mieloma de los previamente descritos y alto riesgo para aquellos que tengan dos o más, o que tengan historia previa de trombosis venosa profunda.6 Habiendo categorizado el riesgo trombótico y descartado el riesgo de sangrado (presencia de trombocitopenia, insuficiencia renal, insuficiencia hepática, hemofilia o hipertensión arterial sistémica) de nuestro paciente, las medidas a tomar serán las que se muestran en la figura 1.

Figura 1 Riesgo trombótico.Dx: diagnóstico; MM: mieloma múltiple; QT: quimioterapia; FR: factor de riesgo; ASA: ácido acetilsalicílico; HBPM: heparina de bajo peso molecular.
En el caso de los pacientes con insuficiencia renal con tasa de filtrado glomerular < 30 ml, se deben considerar ajustes en la dosis o cambio a otro tipo de heparina.
Los procedimientos quirúrgicos suelen ser eventos comunes en pacientes con cáncer, así como las hospitalizaciones prolongadas por toxicidad medicamentosa u otras complicaciones. Durante los periodos de hospitalización se recomienda el uso de HBPM como profilaxis a dosis plena. En caso de que se requiera un procedimiento quirúrgico, las HBPM son también la opción que considerar sobre otros agentes.7
En lo que respecta al manejo de trombosis demostradas en estos pacientes, su manejo dependerá del sitio de trombosis, el grado y la severidad de esta; además, se deberán adecuar las normativas y guías internaciones para cada tipo de trombosis, de ser posible se deberá interconsultar a un experto en trombosis y coagulación. En la tabla 3 se enumeran algunas de las intervenciones recomendadas por diversas guías y series internacionales.
Tabla 3 Tratamiento anticoagulante en el mieloma múltiple
| Tratamiento | Días | Duración |
|---|---|---|
| HBPM 1 mg/kg cada 12 h | 21 días inicial, posterior cambio a AVK, ACD o HBPM (dosis profiláctica) | 4 a 6 meses* |
| AVK | INR 2-3 | 4-6 meses* |
| ACD | No hay evidencia | 4-6 meses* |
*Individualizado al riesgo de cada paciente.
HBPM: heparinas de bajo peso molecular; AVK: antagonistas de la vitamina K; ACD: anticoagulantes directos; INR: international normalized ratio.

