










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkGaceta médica de México
On-line version ISSN 2696-1288Print version ISSN 0016-3813
Gac. Méd. Méx vol.141 n.6 Ciudad de México Nov./Dec. 2005
Caso clínico
Recién nacido con masa mediastinal: diagnóstico temprano
Early diagnosis of a newborn with a mediastinal mass
Salomón Sergio Flores–Hernández,ª* Héctor Ahumada Mendoza,b Blanca Lilia Santana–Montero,c y María de Lourdes González Floresc
ª Hospital Ángeles del Pedregal, México, D. F.
b Servicio de Radiología, Instituto Nacional de Pediatría, México, D. F.
c Servicio de Neurocirugía, Hospital Infantil de México, México, D. F.
d Servicio de Anestesiología, Instituto Nacional de Pediatría, México, D. F., México
*Correspondencia y solicitud de sobretiros:
Dr. Salomón Sergio Flores Hernández.
Esmeralda 9, Col. Valle Escondido, Delegación Tlalpan,
14600 México, D. F.Tel: 5568–8732.
Correo electrónico: ssfloreshdez@yahoo.com.mx
Recibido en su versión modificada: 6 de enero de 2005
Aceptado: 7 de enero de 2005
Resumen
El neuroblastoma es un tumor embrionario que evoluciona a partir de las células de la cresta neural. Esta neoplasia puede originarse en cualquier sitio del sistema nervioso simpático, incluyendo el cerebro, la región cervical, el mediastino posterior, los ganglios simpáticos paraaórticos, la pelvis y la médula de las glándulas suprarrenales. La presentación en la etapa neonatal es rara y en el diagnóstico diferencial deben incluirse las malformaciones congénitas broncopulmonares, neumonías, atelectasias, etc. Se presenta el caso de un recién nacido con neuroblastoma mediastinal, que ejemplifica cómo hacer el abordaje clínico de un paciente con imagen radiológica de masa torácica.
Palabras clave: Neuroblastoma, recién nacido, mediastino.
Summary
Neuroblastoma is an embryonal tumour that evolves from the neural crest cell. This neoplasm may arise at any site in the sympathetic nervous system, including the brain, the cervical region, the posterior mediastinum, the para aortic sympathetic ganglia, the pelvis, and the adrenal medulla. The clinical presentation in neonatal age is rare and the differential diagnosis includes congenital lung malformations, pneumoniae, atelectasia, etc. The case of a newborn with a mediastinal neuroblastoma is presented. This case illustrates how a patient with an X ray image compatible with a thoracic tumor should be studied.
Key words: Neuroblastoma, newborn, mediastinum.
La presencia de opacidad en el tórax diagnosticada en el período neonatal por estudios de imagen plantea la necesidad de descartar en orden de frecuencia las siguientes entidades nosológicas: atelectasia, neumonía y malformaciones congénitas broncopulmonares (del tipo del secuestro pulmonar, quiste neuroentérico, quiste broncogénico, malformación adenomatoidea quística congénita pulmonar, teratoma y otros tumores mediastinales).1
Cuando la localización es en el mediastino posterior se debe considerar la posibilidad de un tumor de origen neurogénico.2 Sin embargo, el médico pediatra o neonatólogo no está acostumbrado a considerar esta patología, ya que se presenta generalmente en pacientes mayores de un año de edad. En este artículo se revisa brevemente esta alteración con la presentación y discusión de un caso clínico.
Caso clínico
El caso corresponde a una recién nacida de un día de edad, producto de una mujer de 33 años de edad, que cursó con embarazo complicado con preeclampsia durante el tercer trimestre. La paciente nació por cesárea por sufrimiento fetal agudo. Presentó un Apgar al nacimiento de 6/8 y SA de 6. El líquido amniótico tenía características meconiales, por lo que se practicó laringoscopia directa que fue negativa, requiriendo presión positiva intermitente. Presentó un paro ca rd i o rres pira–torio que revirtió con maniobras. Permaneció con ventilación mecánica y se trasladó al Instituto Nacional de Pediatría a las 24 horas de nacida.
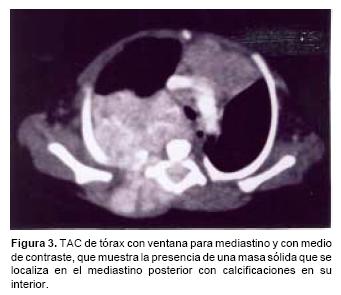
A su ingreso se encontró una paciente con intubación orotraqueal, con dificultad respiratoria (aleteo nasal, tiros intercostales, retracción xifoidea) y hepatoesplenomegalia. Sus signos vitales al ingreso fueron: FC 142, FR 60, TA 91/54 mmHg, TAM 65 y tem 37° C, con un peso de 3,200 g y una talla de 49 cm. Los exámenes de laboratorio informaron gasometría con acidosis respiratoria e hipoxemia. La biometría hemática fue normal al igual que la QS y los ES. La radiografía de tórax mostró una opacidad en el hemitórax derecho, ovoidea, de aproximadamente 8x6 cm con desplazamiento de las estructuras mediastinales hacia la izquierda. Los espacios intercostales en T2–T4 estaban abiertos y el espacio intersomático en T3–T4 estaba incrementado, con adelgazamiento de los arcos costales posteriores en T3 y T4 (Figura 1A). Con estos datos se sospechó la presencia de una masa mediastinal y se solicitó radiografía lateral, la cual confirmó la presencia de una masa localizada en el mediastino posterior, con ensanchamiento del canal raquídeo en T5 y ensanchamiento del agujero de conjunción de T5–T6 (Figura 1B). Se confirmó la presencia de una masa dependiente del mediastino posterior cuyo origen probablemente fuera neurogénico, considerando como primera posibilidad neuroblastoma, de acuerdo con la edad de la paciente. Se realizó ultrasonido torácico y abdominal en los que se identificó un tumor sólido con calcificaciones puntiformes y con tendencia a confluir, el cual involucraba al mediastino posterior, el canal raquídeo y tejidos blandos de la cara posterior del hemitórax derecho (Figura 2A y 2B). En el abdomen había hepatomegalia, con algunas imágenes hipoecoicas que sugerían actividad tumoral. La tomografía computada de tórax confirmó el origen mediastinal de la lesión, las calcificaciones, el involucro y la extensión de la lesión en el canal raquídeo y la afección de los tejidos blandos (Figura 3). La tomografía de abdomen confirmó el compromiso hepático y descartó la presencia de lesiones retroperitoneales.
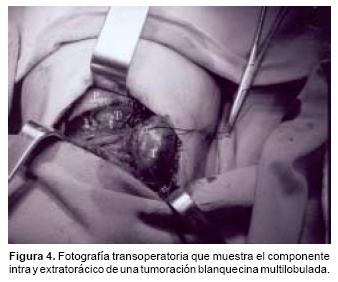
Con los datos descritos se consideró como primera posibilidad diagnóstica un neuroblastoma primario de mediastino, practicándose una toracotomía posterolateral derecha con toma de biopsia incisional de la lesión. Se encontró una lesión con componente extratorácico e intratorácico de aproximadamente 8x6 cm de apariencia cerebroide, de consistencia blanda, fácilmente sangrante y color blanquecino (Figura 4). El estudio histopatológico confirmó el diagnóstico de neuroblastoma indiferenciado (Figuras 5A y 5B). Los estudios de extensión mostraron actividad tumoral en la médula ósea, hígado, arcos costales y columna. Con base en lo anterior se estadificó como estadio IV.
Se decidió iniciar tratamiento con ciclofosfamida, adriamicina y etopósidos (protocolo de St. Jude). Un año después de haber recibido ocho ciclos de quimioterapia, la paciente se encuentra con respuesta favorable y reducción de aproximadamente 95% del volumen tumoral total (Figura 6).

Discusión
El neuroblastoma es un tumor embrionario que evoluciona a partir de las células de la cresta neural. Esta neoplasia puede originarse en cualquier sitio del sistema nervioso simpático incluyendo el cerebro, la región cervical, el mediastino posterior, ganglios simpáticos para–aórticos, la pelvis y la médula de la glándula suprarrenal.3–4 El neuroblastoma es el segundo tumor sólido más común en la niñez, siendo sólo superado por los tumores del sistema nervioso central. Este tumor comprende 7–14% de todas las neoplasias malignas en los niños y es la neoplasia maligna abdominal más común en el período neonatal. Alrededor de 25–50% de todos los pacientes con neuroblastoma son diagnosticados en el primer año de vida.3–4 El tumor es ligeramente más común en los varones que en las mujeres (1.2:1). El neuroblastoma familiar ocurre en gemelos y está asociado con tumores múltiples. En cuanto a su localización, 25–35% se originan en la médula suprarrenal, en segundo lugar en la región paravertebral del mediastino posterior, después en la región paravertebral del abdomen y el resto ocurre en múltiples sitios: pelvis, cuello y cerebro. Su tamaño es variable, desde pequeños nódulos en lesiones in situ a grandes masas con más de 1 kg de peso; la extensión metastásica puede ocurrir por un patrón linfático o hematógeno y aun las lesiones in situ pueden invadir estructuras como el riñón, la vena cava y la vena renal.3–4
El neuroblastoma ha sido raramente detectado como un hallazgo en el ultrasonido fetal.5 Sin embargo, cuando esto ocurre, los tumores tienen generalmente un curso favorable, con múltiples indicadores pronósticos tales como estadio bajo de la enfermedad, marcadores biológicos incluyendo contenido de DNA celular y morfología histológica favorables.6
Los signos y síntomas reflejan la localización de la enfermedad primaria, regional y metastásica. Cuando se origina en el abdomen hay malestar general, masa abdominal fija y dura, y datos de compresión en riñón, vejiga, intestino y otros órganos. Los tumores torácicos primarios se diagnostican incidentalmente cuando se realizan radiografías de tórax; ocasionalmente hay datos de obstrucción mecánica de la vena cava superior.7–8 Los tumores paraespinales pueden extenderse dentro del canal medular y cuerpos vertebrales y causar síntomas de compresión radicular o medular (dolor radicular, paraplejía y disfunción vesical). Por metástasis se pueden observar proptosis, equimosis periorbitaria, manifestaciones hematológicas como hemorragia y trombosis; la invasión a la piel se observa únicamente en niños en estadio IV–S de Evans y se caracteriza por nódulos, que al presionarlos provocan episodios de hipertensión arterial sistémica.
El neuroblastoma puede estar asociado con algunas manifestaciones clínicas no comunes, como "ojos de panda" debido a metástasis orbitarias, opsoclonus/nistagmus (el síndrome del "ojo danzante") el cual está probablemente relacionado a un fenómeno autoinmune, diarrea acuosa debido a la secreción del polipéptido intestinal vasoactivo (VIP), síndrome de Horner en asociación con heterocromía como resultado de un tumor primario originado en el ganglio estrellado cervical, síndrome de dificultad respiratoria causado por una masa ocupativa en mediastino, y hemiplejía relacionada a extensión tumoral a través del foramen intervertebral, resultando en compresión extradural y masa pélvica, lo que ocasiona compresión vascular y vesical.
La evaluación radiológica incluye radiografía de tórax, radiografía de abdomen de pie y acostado, tomografía computada del sitio del tumor primario (cuello, mediastino, abdomen, pelvis) o resonancia magnética nuclear; esta última tiene una especificidad y sensibilidad elevadas. Existen informes recientes de que las reconstrucciones tridimensionales de tomografías computadas helicoidales proporcionan información muy útil que permite evaluar la relación del tumor con los grandes vasos sanguíneos, así como su resecabilidad. En el lactante con un tumor paraespinal, la resonancia magnética nuclear es el estudio de mayor utilidad para evaluar la extensión extradural del tumor y puede documentar compromiso óseo y extensión intraespinal. El aspirado de médula ósea y la biopsia de hueso pueden mostrar rosetas de neuroblastos. El gammagrama óseo con Tc99 y metayodobencilguanidina permiten detectar afección ósea.9 El tumor primario muestra frecuentemente áreas de calcificaciones en las radiografías simples, que se confirman con la tomografía computada. Se pueden determinar en orina concentraciones de catecolaminas, ácido vanillilmandélico y ácido homovanílico. Se deben determinar en suero enolasa neurona–específica10 y concentraciones de ferritina. En pacientes con metástasis hepáticas deben obtenerse determinaciones de tiempos de coagulación y cuenta plaquetaria. Una biometría hemática completa es necesaria para detectar la presencia de anemia.
Los criterios de estadificación11 para el neuroblastoma han sufrido modificaciones en los últimos años y varían de acuerdo al tipo de clasificación utilizados por diversos grupos: la clasificación de Evans para el Grupo de Cáncer en Niños (CCG), la clasificación de St. Jude para el Grupo de Oncología Pediátrica (POG), el Sistema Internacional para la Estadificación del Neuroblastoma, etc. El Sistema Internacional para la Estadificación del Neuroblastoma, la cual es la clasificación más utilizada, incluye 4 estadios:
1. Tumor localizado confinado al área de origen, excisión completa con o sin tumor residual microscópico.
2A. tumor unilateral con resección incompleta, ganglios linfáticos ipsilaterales y contralaterales negativos.
2B. Tumor unilateral con resección completa o incompleta, ganglios ipsilaterales positivos y ganglios linfáticos contralaterales negativos.
3. Tumor que rebasa la línea media con o sin afección de ganglios linfáticos, o tumor unilateral con afección de ganglios linfáticos contralaterales, o tumor de línea media con afección bilateral de ganglios linfáticos,
4.Diseminación del tumor a ganglios linfáticos distantes, hueso, médula ósea, hígado y/u otros órganos,
4–S. Tumor primario localizado como está definido para la etapa 1 ó 2, con diseminación limitada a hígado, piel y/o médula ósea.
Existen diversos marcadores pronósticos:6
1. La edad, generalmente el pronóstico es mejor en menores de 1 año de edad.
2. El sitio del tumor primario, tiene peor pronóstico el neuroblastoma abdominal
3. La estadificación clínica
4. Las características histológicas; según la clasificación de Shimada, la histología favorable son aquellos tumores con estroma rico bien diferenciado.
5. La amplificación del oncogen N–myc en pacientes con neuroblastoma correlaciona inversamente con la edad, la resecabilidad y directamente con el pronóstico.
Hallazgos anatomopatológicos
En la muestra enviada a patología se observó una lesión neoplásica que infiltraba tejidos blandos; estaba formada por células neoplásicas que se distribuían formando cordones y nidos separados por finos septos de tejido fibroso. Las células neoplásicas fueron de aspecto primitivo, de tamaño pequeño, citoplasma escaso, núcleos redondos y de cromatina fina y granular. No se identificaron mitosis ni células ganglionares.
La inmunohistoquímica fue positiva para enolasa neurona específica y para sinaptofisina. En el estudio ultraestructural se encontraron células pequeñas, algunas con neurofibrillas en el citoplasma, todas con cantidad variable de gránulos neurosecretores sin membrana. Todos estos datos hacen el diagnóstico de neuroblastoma indiferenciado. En los frotis examinados del aspirado de médula ósea se observaron elementos de la serie mieloide, y linfoide sin alteraciones histológicas; mezclados entre éstos se encontraban pequeños grupos de células neoplásicas pequeñas azules y cohesivas que formaban rosetas. Las células tenían citoplasma escaso y núcleos grandes de cromatina fina. La inmunohistoquímica fue positiva para enolasa neurona específica y sinaptofisina en las células neoplásicas.
En resumen, la presencia de una lesión ocupativa en el tórax debe hacer pensar en varias posibilidades; en primer lugar en lesiones benignas del tipo de las malformaciones congénitas broncopulmonares (secuestro pulmonar, quiste broncogénico, duplicación intestinal). También deben considerarse patologías malignas como son los tumores de origen neurogénico, específicamente el neuroblastoma. Algunos datos clínicos podrían ayudar a establecer el diagnóstico como son la presencia de diarrea y eventos de hipertensión arterial sistémica en el caso del neuroblastoma. Los estudios que realmente van a permitir el diagnóstico diferencial son los estudios de imagen, en primer lugar la radiografía simple de tórax, en la que en el caso del neuroblastoma pueden observarse calcificaciones, siendo el de mayor utilidad la tomografía axial computada de tórax, que va a permitir diferenciar entre una lesión quística y una sólida, así como definir la localización topográfica con gran precisión y su relación con estructuras vasculares de gran importancia para el abordaje quirúrgico.
Referencias
1. Chest and respiratory system. En: Essentials of pediatric radiology. Burton EM, Brody AS(eds) Thieme primera edición New York, 1999. p. 273–314. [ Links ]
2. Saenz NC, Schnitzer JJ, Eraklis AE, Hendren WH, Grier HE, Macklis RM, Shamberger RC. Posterior mediastinal masses. J Pediatr Surg. 1993; 28(2) 172–176. [ Links ]
3. Grosfeld JL. Neuroblastoma. En: Pediatric surgery. O' Neill JA, Rowe MI, Grosfeld JL, Fonkalsrud EW, Coran AG (eds) Mosby 5a. edición St. Louis, 1998. p. 405–419. [ Links ]
4. Grosfeld J. Neuroblastoma. En: Neonatal tumours. Puri P (ed) Springer la. edición London, 1996. p. 29–42. [ Links ]
5. Acharya S, Jayabose S, Kogan SJ, Tugal O, Beneck D, et al. Prenatally diagnosed neuroblastoma. Cancer 1997; 80(2):304–310. [ Links ]
6. Castleberry RP. Predicting outcome in neuroblastoma. N Engl J Med 1999; 340(25): 1992–1993. [ Links ]
7. Suita S, Tajiri T, Sera Y, Takamatsu H, Mizote H, et al. The characteristics of mediastinal neuroblastoma. Eur J Pediatr Surg 2000; 10(6):353–359. [ Links ]
8. Eklof O, Friis M. Thoracic neuroblastoma of the neonate. Acta Radiol Diagn (Stockh). 1985; 26(4) :441 –446. [ Links ]
9. Shulkin BL, Shapiro B. Current concepts on the diagnostic use of MIBG in children. J Nucl Med 1998;39(4):679–688. [ Links ]
10. Massaron S, Seregni E, Luksch R, Casanova M, Botti C, et al. Neuron–specific enolase evaluation in patients with neuroblastoma. Tumour Biol 1998; 19(4):261–268. [ Links ]
11. Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, et al. Terminology and morphologic criteria of neuroblastic tumors: recommendations by the Intemational Neuroblastoma Pathology Committee. Cancer 1999; 86(2):349–363. [ Links ]

