










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkGaceta médica de México
versão On-line ISSN 2696-1288versão impressa ISSN 0016-3813
Gac. Méd. Méx vol.141 no.3 Ciudad de México Mai./Jun. 2005
Caso clínico
Deficiencia congénita de proteína C. Informe de un caso
Protein C congenital deficiency. A case report
María de Lourdes Lemus–Varela,ª* José de Jesús Arriaga–Dávilaª y Martha Patricia Salinas–Lópezª
* Correspondencia y solicitud de sobretiros:
María de Lourdes Lemus Varela
Departamento de Neonatología del Hospital de Pediatría del C.M.N.O., IM.M.S.
Belisario Domínguez 735 Sector Reforma,
C.P. 44340, Guadalajara Jalisco, México. Tel.: 36683000; ext.: 31729 y 31730.
Correo electrónico: lulalemus@hotmail.com
ª Departamento de Neonatología, Hospital de Pediatría del Centro Médico Nacional de Occidente, lMSS, Guadalajara, Jal., México
Recibido en su versión modificada: 21 de junio de 2004
Aceptado: 13 de agosto de 2004
Resumen
La proteína C (PC) es una proteína plasmática que se sintetiza en el hígado con el apoyo de la vitamina K y regula la formación de trombina y consecuentemente la prevención de una trombosis. Se presenta el caso de un recién nacido masculino, con cambio de coloración en primer dedo del pie derecho, y 4 h después cianosis hasta nivel maleolar. A su ingreso se encontró con palidez generalizada, taquicárdico y con lesión necrótica en pie derecho. Inicialmente se sospechó proceso séptico por lo cual se manejó con Cefotaxima, Vancomicina y heparina. Presentó plaquetopenia 70,000mm3, tiempo de tromboplastina 16/12 seg. y tiempo de tromboplastina parcial de 58/29 seg., con funcionalidad de PC del 20% y proteína S de 100%. A pesar de mostrar evolución favorable y una recuperación parcial de la zona afectada, requirió amputación infratuberocitaria, además de manejo con enoxaheparina que posteriormente se cambió por acenocumarina, poco después del año de edad, se colocó prótesis. Se discute la conveniencia de continuar con estudios que apoyen el uso de anticuerpos monoclonales de PC a fin de dar el tratamiento sustitutivo de base y mejorar la calidad de vida de pacientes con deficiencia congénita de la misma.
Palabras clave: Proteína C, deficiencia congénita, trombosis
Summary
Protein C is a plasmatic protein that is synthesized by the liver with the help of vitamin K. It regulates thrombin formation and consequently prevents thrombosis. We present a case of a newborn male with change in the color of the right foot index finger who after 4 h showed cyanosis that reached malleolus level. Upon admission we observed generalized pallor, tachycardia and a necrotic lesion in the right foot. We suspected a septic process and thus administered cefotaxime, vancomycin and heparin. Platelet levels were 70,000mm3, thromboplastin 16/12 sec., partial thromboplastin 5829 sec. PC functionality 20% and protein S 100%. Even though the patient evolved favourably and showed partial recovery, an intratuberous amputation was needed. One year later a prosthesis was fitted. We need to carry out studies that support the use of PC monoclonal antibodies in order to offer better baseline treatment to patients with PC congenital deficiency and improve their quality of life.
Key words: Protein C, congenital deficiency, thrombosis
Introducción
La proteína C (PC) es una proteína plasmática que se sintetiza en el hígado con el apoyo de la vitamina K. Su principal acción es regular la formación de trombina y consecuentemente la prevención de una trombosis.1,2 La acción inicia cuando la trombina se une a la trombomodulina (TM), que se encuentra en la superficie de las células endoteliales, esta unión activa a la PC y simultáneamente bloquea la capacidad de la trombina de catalizar la formación de fibrina, activación del factor XIII, activación plaquetaria y retroalimentación negativa para la activación de los factores de la coagulación.3
En algunos vasos sanguíneos, la activación de la PC puede ser incrementada, por la presencia de un receptor endotelial (EPCR por sus iniciales en inglés), el cual se une a la PC y a la proteína C activada, con igual afinidad, y favorece la funcionalidad de la misma.4 Una vez activada la PC, se une a la proteína S (PS) en la superficie de las células endoteliales y este complejo inactiva a los factores Va y Vllla, que son esenciales para la formación final de trombina, lo cual a su vez también limita la nueva activación de la PC (Figura 1).2

La respuesta de la trombina es controlada por inhibidores plasmáticos. En el caso del complejo de activación de la PC, la trombina unida a la TM es inhibida por la antitrombina (ATT) y este complejo de trombina ATT es rápidamente disociado de la TM para producir un cofactor anticoagulante activo (Figura 1).2
La vida media del complejo trombina–trombomodulina a concentraciones fisiológicas, es de sólo algunos segundos.5 Mientras que la vida media de la PC activada es poco más de 15 minutos, y su inactivación en este caso está mediada por los inhibidores de la PC como antitripsina y macroglobulina.6
El déficit congénito de PC se describe como un rasgo autosómico dominante, con una incidencia de uno por cada 16 mil recién nacidos vivos y se divide en dos tipos: el más frecuente que es el tipo 1, en el que los pacientes tienen tanto disminución de la PC como de su actividad y la tipo II en la cual el paciente tiene una función alterada de la PC, pero su concentración plasmática es normal.7,8 Los pacientes homocigotos con deficiencia de PC se presentan clínicamente con un cuadro de púrpura fulminante en los primeros días de vida. A menudo presentan eventos trombóticos a nivel de sistema nervioso central, ocular, trombosis de grandes vasos y datos de coagulación intravascular diseminada (CID) y pueden morir por complicaciones de una púrpura fulminante, durante el primer mes de vida. Los neonatos heterocigotos presentan lesiones trombóticas a nivel cutáneo, renal o en vena umbilical.8 El criterio diagnóstico en la deficiencia congénita de PC en los pacientes homocigotos incluye disminución persistente o ausencia de la concentración de PC y heterocigocidad por deficiencia de PC en ambos padres.8 Las concentraciones normales de PC en recién nacidos pretérmino son de 0.28 a 0.37 u/mL y en recién nacidos a término de 0.35 a 0.43 u/mL.8
En forma general el tratamiento consiste en dar el apoyo sintomático con plasma fresco, y terapia anticoagulante con Heparina y sus derivados. En fechas recientes se ha propuesto el tratamiento sustitutivo con el uso de PC monoclonal.10
En este informe describimos un caso clínico de deficiencia congénita de PC.
Caso clínico
Paciente masculino de 12 días de edad posnatal, producto de la cuarta gestación de madre de 28 años, sana y padre de 31 años, referido sano, se informó embarazo normoevolutivo con control prenatal adecuado, que culmina en parto eutócico obteniendo producto masculino, con peso de 3200 g, Apgar: 8–9, egresado a su domicilio con la madre poco después de 24 h de su nacimiento.
Inició su padecimiento a los 10 días de vida, la madre nota cambio de coloración en el primer dedo del pie derecho; 4 h después se observó la presencia de cianosis hasta nivel maleolar (marca del calcetín), motivo por el cual acudió a su hospital general, que deriva a nuestra sala.
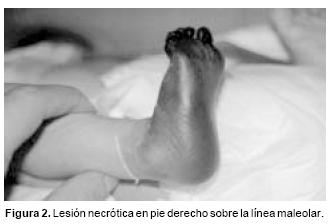
A su ingreso se encontró con palidez generalizada, taquicárdico y con presencia de lesión necrótica en pie derecho sobre la línea maleolar e hipotermia del área de lesión (Figura 2). Por la evolución y hallazgos clínicos y de laboratorio se consideró un proceso séptico agregado, con trombosis de pie derecho, probablemente secundaria a CID. Se inició manejo con doble esquema de antimicrobiano (Cefotaxima y Vancomicina), además de heparina a razón de 30 u/kg/día.
Se solicitó valoración por el servicio de angiología, que diagnosticó trombosis de pie derecho de etiología a determinar y sugiriendo continuar con heparina. En los servicios de traumatología y ortopedia consideraron, por la gravedad de la lesión, la posibilidad de amputación y en hematología sospecharon una alteración en la coagulación, probablemente deficiencia de PC. Los resultados de laboratorio iniciales fueron: biometría hemática con hemoglobina y hematócrito normales, leucocitos y diferencial también en márgenes normales, pero con plaquetas de 70.000mm3, tiempo de protrombina (TP) de 16/12 seg., y tiempo de tromboplastina parcial (TTP) de 58/29 seg., iniciándose manejo con plasma fresco.
Así mismo se valoró por el servicio de oftalmología, a fin de descartar hemorragia de cuerpo vítreo, lo cual es una manifestación a nivel ocular frecuente en pacientes con deficiencia de PC;12 esta posibilidad fue descartada.
Tres días más tarde se obtuvo el resultado de proteína C, con funcionalidad del 20% y proteína S al 100%. Se continuó con el manejo médico establecido con heparina, se suspendió esquema de antibióticos, y a pesar de mostrar una evolución favorable, finalmente requirió de amputación infratuberocitaria derecha, la cual se realizó sin inconvenientes.
Cinco días después fue egresado del hospital con heparina de bajo peso molecular (enoxaheparina) subcutánea cada 24 h y control por consulta externa multidisciplinaria.
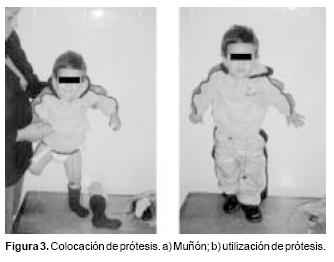
Acudió a su primera cita postquirúrgica a los 21 días de su egreso, encontrándose en buenas condiciones generales, con muñón de buena coloración, sin datos de trombosis a otro nivel, con un TP de 19/13 seg. Se decidió suspender la heparina de bajo peso molecular, e iniciar tratamiento con acenocumarina a 0.25 mg/kg. Así mismo fue valorado por medicina física iniciando terapia de rehabilitación. Ocho meses después se colocó prótesis (Figura 3a y 3b). En la actualidad continúa su tratamiento con acenocumarina, además con apoyo de rehabilitación, con lo que ha tenido evolución favorable. En su última evaluación presentó, un TP de 12 /11 seg por lo que se incrementó dosis de acenocumarina a 0.50 mg/kg y se inició fisioterapia para utilización de prótesis (Figura 3a y 3b).
Comentario
El paciente tuvo manifestaciones clínicas dentro de los primeros 15 días de vida con cambios de coloración muy limitados al extremo distal de miembro pélvico derecho, a su ingreso, por apoyo clínico y de laboratorio se sospechó un proceso séptico, el cual fue descartado debido a que por la evolución clínica y de laboratorio, especialmente por los tiempos de coagulación prolongados se pensó en la posibilidad de cursar con una deficiencia congénita de PC, como se menciona en la literatura la edad de inicio y las características clínicas y de laboratorio, son propias de una deficiencia de PC tipo 1, además de tratarse muy probablemente de un paciente heterocigoto.8
En un estudio realizado por Ruíz–Argüelles y cols.9 en 102 pacientes mexicanos con datos clínicos de trombosis, encontró que 39% de ellos tenían resistencia a la PC, sólo 5% tenía deficiencia de PC y 2 % deficiencia de PS.9
Los pacientes homocigotos presentan a menudo una púrpura fulminante que ocasiona la muerte temprana,11 mientras que los pacientes heterocigotos tienen un curso más benigno con manifestaciones cutáneas, renales y oculares, dentro de las cuales la más frecuente es una hemorragia del cuerno vítreo, seguido por trastornos retinianos.12
En la actualidad el tratamiento en un inicio es sintomático, con plasma fresco, Heparina y anticoagulantes orales.8 Sin embargo se han descrito nuevas y alentadoras expectativas de manejo, como el uso de anticuerpos monoclonales de PC, y se describe en algunos estudios la mejoría en la concentración de PC al cabo de 20 días, con una disminución importante de la sintomatología, y sin requerir en ocasiones de tratamiento coadyuvante con anticoagulante, o bien con disminución en la dosis del mismo. Al parecer no se han informado efectos colaterales con esta modalidad y se ha logrado disminuir de manera importante la estancia intrahospitalaria y especialmente la calidad de vida de estos pacientes.10
La evolución clínica y de laboratorio del presente caso, fue la del tipo más frecuente de deficiencia de PC, con una actividad del 20%, y aunque fue necesario amputar una extremidad, el paciente en la actualidad tiene una evolución estable, y un curso benigno. Con manejo a base de anticoagulantes del tipo de la acenocumarina.
De lo anterior se concluye, que dada la poca frecuencia de esta entidad, es necesario tomarla en cuenta ante un paciente con un cuadro clínico sugestivo de trombosis, a fin de prevenir las secuelas que pueden originar una discapacidad de grado variable o incluso la muerte.
El diagnóstico temprano y tratamiento oportuno están relacionados directamente con la evolución, sin embargo en nuestro país contamos hasta el momento únicamente con la terapia de apoyo, y el manejo anticoagulante tradicional, como heparina y sus derivados, o bien cumarínicos. Con los efectos colaterales que éstos pueden presentar.
Así pues será conveniente continuar con estudios que apoyen el uso de anticuerpos monoclonales de PC a fin de dar el tratamiento sustitutivo de base, a fin de disminuir las complicaciones, y mejorar sobre todo la calidad de vida en estos pacientes.
Referencias
1. Best Taylor. Bases Fisiológicas de la Práctica Médica. Ed. Panamericana. Buenos Aires, 1986. [ Links ]
2. Andrew M, Brooker LA. Trastornos de la coagulación en los recién nacidos. En tratado de neonatologia de Avery. Taeusch HW, Ballard RA. Harcourt S.A. USA. 2000, PP 1045–1079. [ Links ]
3. Esmon CT, Cell mediated events that control blood coagulation and vascular injury. Annu Rev Cell Biol 1993; 9:1–26 Review [ Links ]
4. Fukudome K, Esmon CT. Identification, cloning and regulation of a novel endothelial cell protein C/ activated protein C receptor. J Biol Chem 1994; 269:26486–26491. [ Links ]
5. Rezaie AR, Cooper ST, Church FC, Esmon CT. Protein C inhibitor is a potent inhibitor of the thrombin–thrombomodulin complex. J Biol Chem 1995; 270:25336–25339. [ Links ]
6. Heeb MJ. España F, Griffin JH. Inhibition and complexation of activated protein C by two major lnhibitors in plasma. Blood 1989; 73:446–454. [ Links ]
7. Montgomery RR, Scott JP. Enfermedades hemorrágicas y trombóticas. En: Tratado de Pediatría de Nelson, (Ed) McGraw–Hill Inc 16a Edición Philadelphia Pennsylvania USA, 2000, pp 1645–1077. [ Links ]
8. Edstrom CS, Christensen RD, Andrew M. Developmental aspects of blood hemostasis and disorders of coagulation and fibrinikysis in the neonatal period. En: Christentsen RD. Hematologic problems of the neonate. WB Saunders. Philadelphia Pennsylvania 1st. edition, 2001, pp 239–271. [ Links ]
9. Ruiz–Arguelles GJ, González–Estrada S, Garces–Eisele J. Primary trombophilia in Mexico: a prospective study. Am J Hematol 1999; 60:1–5. [ Links ]
10. Dreyfus M, Masterson M, David M, Rivard GE. Replacement therapy with a monoclonal antibody purified protein C concentrate in newborns with severe congenital protein C deficiency. Semin thromb hemost.1995; 21:371–381. [ Links ]
11. Pescatore SL. Clinical management of protein C deficiency. Expert Opin Pharmacother. 2001 ;3:431–439. [ Links ]
12. Hattenbach LO, Beeg T, Kreuz W, Zubcov A. Ophthalmic manifestation of congenital protein C deficiency. J AA POS 1999; 3:188–190. [ Links ]

