











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La alcaptonuria es una enfermedad metabólica rara, su etiología está dada por déficit de la enzima oxidasa del ácido homogentísico, dentro de su clínica y evolución es responsable de afectar grandes articulaciones, generando cambios artrósicos secundarios. Es de herencia autosómica recesiva y la mutación para la enzima alterada se encuentra en el cromosoma 3q21q23. Su incidencia es de 1/250,000-1’000,000;1 sin embargo, en República Dominicana y Eslovaquia se ha reportado 1/19,000. El primer caso reportado en la literatura fue por Boedeker en 1854, en 1901 Garrod describe su patrón de herencia, en 1904 Osler describe su clínica y en nuestro país el primer reporte data de 1981 por el Dr. Antonio Iglesias.2
Debido al mecanismo de herencia, los padres de los pacientes que padecen alcaptonuria regularmente serán portadores asintomáticos, lo cual les da una probabilidad de 25% de padecer la enfermedad. Sus manifestaciones clínicas inician desde temprana edad, siendo el principal signo el cambio en la coloración de la orina, la cual al exponerse al sol se torna más oscura, asimismo están descritas manchas negras en los pañales de los infantes; sin embargo, ya que es el único signo, es difícil su diagnóstico a edad temprana. Alrededor de la tercera o cuarta década de la vida inicia su clínica más notable, evidenciándose ocronosis (coloración negra/azul del tejido conectivo) y poliartralgias, comprometiendo las articulaciones periféricas y el esqueleto axial: la columna con cambios degenerativos generando fibrosis de los discos intervertebrales, la articulación de la rodilla > 60% de los casos, el hombro en 43% de los casos y las caderas en 35% de los casos. Las pequeñas articulaciones como las manos y los pies también son afectadas, pero en menor proporción. Se ha descrito adicionalmente dado el compromiso del tejido conectivo rupturas espontáneas del tendón patelar y del tendón de Aquiles.3 Teniendo en cuenta que es una enfermedad metabólica, los pacientes pueden presentar episodios de litiasis renal por acúmulo del ácido homogentísico y afectaciones valvulares cardíacas con calcificación, éstas especialmente después de los 50 años de edad.
En cuanto a su diagnóstico, se debe basar en la anamnesis con un interrogatorio adecuado, teniendo presentes las características de la orina, la ocronosis (especialmente en pabellón auricular y las escleras) (Figuras 1 y 2) y el dolor en las grandes articulaciones. Su diagnóstico confirmatorio se realiza mediante la detección de HGA en orina, por espectrometría de masa de cromatografía de gas o mediante análisis genético. En los diagnósticos diferenciales está la artritis reumatoidea, espondilitis anquilosante, enfermedad de Paget y tumores óseos.4


Haciendo referencia al tratamiento, lo ideal sería realizar la suplencia de la enzima; sin embargo, actualmente no existe un manejo efectivo para la misma, por lo cual su tratamiento se limita a tratar los síntomas, en el momento que las poliartrosis son avanzadas, la artroplastía es una opción con excelentes resultados.4
En esta serie de casos se presenta a un hombre de 55 años con un compromiso de todas las grandes articulaciones y una mujer de 58 años con artrosis bilateral de la cadera y hombro.
Serie de casos
Caso número 1. Hombre de 55 años sin antecedentes patológicos de importancia, médico de profesión, mestizo, quien refiere que a los 30 años de edad durante actividad deportiva presenta trauma rotacional en rodilla derecha, es intervenido en el momento, mediante artroscopía por sospecha de lesión menisco medial, realizan desbridamiento y como hallazgo encontraron coloración negra en superficie articular, indicando continuar manejo sintomático y observacional. En 2005 el paciente consulta por dolor lumbar severo, pérdida progresiva de la fuerza en miembros inferiores, estudios imagenológicos documentan espondiloartropatía y realizan manejo con laminectomía posterior, artrodesis lumbar y torácica. Seguido a procedimiento quirúrgico de columna, en 2006 el paciente consulta nuevamente por síntomas mecánicos en rodillas, llevándolo a nueva artroscopía con remodelación meniscal, se hace manejo de dolor y se documentan cambios artrósicos. En 2007 por persistencia de síntomas y hallazgos, se le realiza reemplazo total de rodilla izquierda. En el año 2009 le es practicado un reemplazo total de rodilla derecha. Procedimientos hasta la fecha con adecuada evolución y sin aparición de nuevos síntomas para esta articulación.
Para 2014, el caso presenta dolor severo a nivel de hombro izquierdo, donde realizan reemplazo con prótesis anatómica. Durante el año 2017, manifiesta que durante la marcha presenta sensación de patada a nivel de gastrocnemio izquierdo asociada a posterior dolor, al examen físico con signo de Thompson positivo y brecha a nivel del tendón de Aquiles, por lo que se considera una ruptura espontánea del Aquiles, un mes después presenta ruptura espontánea del tendón de Aquiles derecho. En ambos se indicó manejo conservador con adecuada respuesta. En el 2020 presenta dolor y cambios inflamatorios en el hombro izquierdo, documentándose infección periprotésica con aislamiento de S. marcescens y P. acnes, manejada con antibioticoterapia y completando esquema, actualmente se encuentra con espaciador de cemento funcional.
En Enero de 2022 al realizar bipedestación presenta sensación de traquido y dolor intenso en cadera derecha, limitando la marcha y requiriendo uso de caminador. Después de evaluación con radiografía de pelvis (Figura 3), es llevado a reemplazo total de cadera derecha (Figura 4). La Figura 5 muestra la coloración negro/azul del acetábulo. Al interrogatorio para el procedimiento quirúrgico, refiere consanguinidad entre abuelos y padres (primos), durante infancia orinas oscuras intermitentes y aumento de la coloración en conjuntivas, aproximadamente hace cinco años más evidente. En la actualidad utiliza un bastón canadiense para apoyo de la marcha. Además de sus manifestaciones óseas y articulares, dentro de sus antecedentes ha presentado tres episodios de litiasis renal, requiriendo intervención quirúrgica y además de episodios recurrentes de pérdidas inmotivadas de piezas dentales.

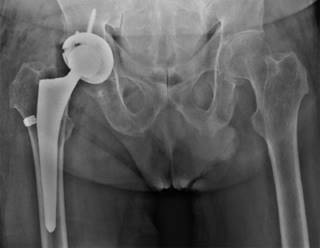
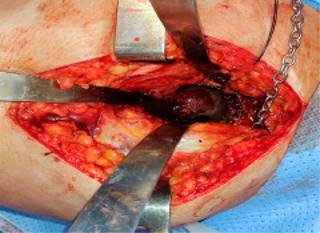
Ante la clínica del paciente y correlacionando sus signos y síntomas, se sospechó patología de origen metabólico, por lo cual se diagnosticó alcaptonuria realizando determinación de ácido homogentísico en orina 24 horas con un valor de 2,100 mg/día (valor < 10 mg/día). La Figura 6 muestra las imágenes histológicas del cartílago articular.
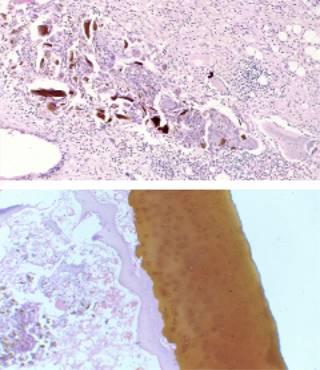
Figura 6: Imágenes histológicas del caso número 1, tinción hematoxilina-eosina: células gigantes pigmentadas y coloración del cartílago articular (imágenes tomadas por la Dra. Ana María Cock, Patología Clínica Las Vegas Grupo Quirónsalud).
Caso número 2. Mujer de 58 años, tecnóloga en sistemas, mestiza, con antecedente personal de lupus eritematoso sistémico, enfermedad renal crónica estadio III, hipertensión arterial e hipotiroidismo. Refiere desde hace 15 años inicio de dolor lumbar, manejado con terapia física, durante crecimiento y desarrollo no presentó síntomas, ni cambios en la coloración en la orina; sin embargo, hacia la adolescencia y adultez, refiere aparición y aumento progresivo de coloración negra en pabellones auriculares y escleras, además deformidad en dedos de las manos. En 2015 manifiesta dolor, el cual genera limitación y presenta cambios artrósicos severos en cadera derecha, síntomas y signos tratados mediante remplazo total de cadera. Para el año 2017, presenta dolor severo en hombros con artrosis avanzada documentada en imágenes, por lo cual le realizan reemplazo bilateral de hombros, con una diferencia de 10 meses entre uno y otro. Durante este procedimiento se evidencia coloración negra de cabeza humeral, sospechando alcaptonuria, razón por la que se realiza biopsia y se remite al servicio de genética, quienes complementan estudios con análisis de ácido homogentísico en sangre, el cual confirma el diagnóstico de alcaptonuria, encontrando la alteración en el gen HGD (variante c.673C>T), el análisis en orina fue negativo con un valor de 3.34 mg/día. Cuatro años después del diagnóstico, la paciente refiere inicio de dolor severo en cadera izquierda con limitación funcional y le es documentada artrosis Tonnis 3 (Figura 7). Por lo que en Febrero de 2022 se realiza reemplazo total de cadera izquierda (Figura 8). Actualmente niega síntomas a nivel de articulaciones operadas.

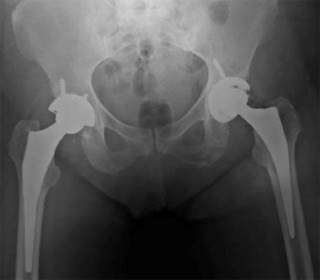
Dentro de los antecedentes ya mencionados, es importante resaltar como antecedente familiar hermana de 66 años de edad, con diagnóstico por genética de alcaptonuria con artropatía ocronótica secundaria, la cual ha recibido manejo con reemplazo de hombro izquierdo, cadera y rodilla bilateral. Además, su hermano de 64 años de edad tiene antecedente quirúrgico de reemplazo bilateral de rodillas, indicado por artrosis, en quien aún no se ha confirmado el diagnóstico con análisis de ácido homogentísico; sin embargo, presenta ocronosis en escleras y pabellones auriculares. Estos familiares han presentado desde la infancia coloración negra intermitente en la orina. Y al remontarse en su historia familiar, se concluye que la paciente y sus dos hermanos son producto de padres con consanguinidad (primos hermanos).
Con el diagnóstico basado en la radiografía simple de pelvis (Figura 7) los casos expuestos fueron llevados a reemplazo total de cadera. Caso número 1 cadera derecha, caso número 2 cadera izquierda. Los dos procedimientos se realizaron mediante abordaje posterolateral por el mismo cirujano, en ambos se tenotomizan los rotadores cortos en su parte tendinosa para posterior reinserción, se realiza capsulotomía longitudinal. En el caso número 1 se identificó intraoperatoriamente una fractura de la parte proximal de la cabeza femoral, la cual podría explicar la sintomatología de inicio agudo del paciente. En el caso número 2 se identificaron cambios artrósicos avanzados, en ambos procedimientos de observa pigmentación de la cabeza femoral y la cavidad acetabular (Figura 9). Los dos casos recibieron manejo con prótesis primaria total de cadera no cementada y dado calidad ósea se aumentó la fijación acetabular con tornillos. Para el postoperatorio inmediato se inició tromboprofilaxis con heparina de bajo peso molecular, se realizó hemoglobina y hematocrito de control sin presentar anemia. Se inició bipedestación y marcha en el primer día postoperatorio con caminador sin complicaciones. Los pacientes fueron dados de alta en el segundo día postoperatorio sin ninguna complicación.

Discusión
Históricamente el número de casos reportados y publicaciones sobre esta patología son reducidos, el primer caso en la literatura data de 1854 por Boedeker, en 1901 Garrod describe el patrón de herencia para la patología, durante 1904 Osler describe su clínica. En Colombia el primer reporte se dio en 1981 por el Dr. Antonio Iglesias en Barranquilla,2 en el cual reporta un caso de un paciente de 57 años con antecedente de reemplazo bilateral de cadera y afectación de pequeñas articulaciones. Para el año 2008 el Dr. Donado reporta caso de espondiloartropatía tratada, posteriormente en 2012 en Bogotá el Dr. Galván reporta un caso de paciente con antecedente de reemplazo de cadera derecha que presenta adicionalmente fractura de cadera izquierda inveterada, para la cual se indica manejo conservador.5,6,7 Hacia el año 2015 el Dr. Fierro documenta el caso de artropatía de hombro manejado con artroscopía8 y en 2017 el último reporte de caso es de un paciente de 12 años de edad, en el cual se realiza el diagnóstico de la enfermedad.9
La escasa bibliografía existente tiene relación con la forma de presentación y enmascaramiento de síntomas que se producen en esta patología, incluso es definida como un síndrome raro producido por la ausencia de la enzima oxidasa homogentísica. Epidemiológicamente su incidencia está dada por 1/250,000 y 1/1’000,000, lo que ratifica la probabilidad de presentar el síndrome, frente a los casos conocidos.
En cuanto a la fisiopatología se debe al depósito del ácido homogentísico en el tejido conectivo por deficiencia de la enzima ácido homogentísico 1,2 dioxigenasa, lo cual evita el desdoblamiento en ácido maleilacetoacético (vía de la tirosina), llevando al acúmulo de benzoquinona y melanina, lo que se atribuye la coloración. En cuanto al daño articular que produce, no se ha descrito la causa clara; sin embargo, se plantean tres hipótesis: si el depósito del ácido actúa como un irritante químico, el daño es causado por radicales libres generados por el benzoquinoacetato o por alteración de la lisil hidroxilasa enzima esencial para mantener enlaces cruzados del tejido conectivo débil, lo cual lo podría debilitar, generando daño articular, mala calidad ósea y pudiendo generar incluso fracturas y patologías.10
La alcaptonuria, al no ser un síndrome de total conocimiento para quienes tratan su sintomatología, se mencionan las características importantes para que así se logre correlacionar con los casos presentados, su alteración genética se encuentra en el brazo largo del cromosoma 3, con patrón de herencia autosómico recesivo, esto conduce a una alteración en el metabolismo del ácido homogentísico, generando un acúmulo tanto en las articulaciones como en el tejido conectivo.11,12 Los pacientes presentan manifestaciones en la orina desde temprana edad con coloración oscura intermitente, siendo quizás este el primer signo, aunque pasa desapercibido haciéndose más notable hacia la tercera o cuarta década de la vida porque es el momento de mayor acumulación en los tejidos blandos, generando la ocronosis y dando sus manifestaciones a nivel articular mediante el dolor, afectando en más de 60% principalmente la columna, seguida de la articulación de la rodilla.
Con relación a la sintomatología, es importante mencionar que en el metabolismo normal del hígado tiene la capacidad suficiente de producir oxidasa de HGA para convertir 1.5 kg de ácido homogentísico al día, dado lo anterior, es necesaria la pérdida de > 99% de la enzima para que un caso sea sintomático. La enfermedad comúnmente es asintomática hasta la tercera o cuarta década de la vida, en donde generalmente la alteración articular comienza a manifestarse y aumenta de manera progresiva requiriendo intervención, hacia la cuarta o quinta década de la vida.
Lo anterior fundamenta el criterio médico para generar la sospecha clínica y realizar su diagnóstico definitivo, mediante la identificación de niveles elevados de ácido homogentísico (> 10 mg/día) en la orina en 24 horas o mediante análisis genético.
El tratamiento idóneo seria sustituir la enzima faltante; sin embargo, en la actualidad no está disponible. Se han implementado diferentes manejos como bifosfonatos, vitamina C, restricción de ingesta proteica, con desenlaces no efectivos. Se ha planteado el uso de la nitisinona, medicamento que inhibe la 4 hidroxifenilpiruvato dioxigenasa (metabolismo de la tirosina), lo cual disminuye la ocronosis, pero no posee impacto directo sobre la progresión de la enfermedad, dejando como única opción hacer manejo sintomático y en caso de cambios artrósicos avanzados el reemplazo articular. Quizás la única opción de cura para la alcaptonuria es el trasplante hepático, lo cual sustituiría el déficit enzimático, pero no se encontró literatura que respalde e indique el manejo aislado con trasplante para este síndrome. Hay escasos reportes de caso en los que la ocronosis desaparece después del trasplante hepático, por lo que la indicación de esta falla hepática es hepatitis y cirrosis, y no la alcaptonuria.
Al día de hoy después de realizar una revisión amplia de la literatura y teniendo en cuenta que la mayoría de estudios son reportes de caso, no se encontró en nuestro país la documentación como la de nuestro caso número 1, en el cual podemos evidenciar el gran compromiso musculoesquelético, debido a la alcaptonuria y las diferentes manifestaciones clínicas amplias, afectando además la pérdida de piezas dentales, litiasis renal, ruptura espontánea de ambos tendones de Aquiles (reportados en la literatura que se pueden dar hasta 20%) y una artropatía ocronótica generalizada, siendo el primer caso documentado en nuestro país en el cual se ha realizado manejo con artroplastía en tres articulaciones periféricas. Se reporta la mejoría temprana de los síntomas y el éxito del reemplazo de cadera en dos casos con artropatía secundaria medido por Harris Hip score, además la supervivencia a largo plazo de las artroplastías de rodilla, sin presentar aflojamiento, lo cual nos indica que la artroplastía es un excelente manejo para dicha patología.
Conclusiones
La alcaptonuria es una enfermedad metabólica poco frecuente que afecta la columna, las grandes articulaciones y el tejido conectivo, en la actualidad no hay un tratamiento específico que permita la resolución de este síndrome, lo cual limita a que su manejo sea paliativo. En caso de presentar compromiso articular severo, las artroplastías son una alternativa para control del dolor y pueden ayudar a mantener la funcionalidad de los pacientes a largo plazo.

