











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Colorectal cancer (CRC) is one of the most prevalent gastrointestinal cancers worldwide, especially in Western and European countries and has been introduced as the most common cancer type in Asia1,2. Hereditary nonpolyposis colorectal cancer (HNPCC) or Lynch syndrome, the most frequent inherited syndrome, is a highly penetrant disease (about 85%) with the autosomal dominant inheritance patterns that account for approximately 5-10% of total CRC cases3. Patients with HNPCC are at a high risk to develop a range of cancers, including colorectal and endometrial cancers as well as extracolonic gastrointestinal, genitourinary, and ovarian and brain cancers4,5. Mutations in MLH1 and MSH2 are considered the major cause of HNPCC, since germline alterations of these genes have been found to be responsible for more than 90% of mutation carrier HNPCC families. According to the database of the International Society for Gastrointestinal Hereditary Tumors (InSiGHT), currently more than 450 different pathogenic mutations have been described in these genes, accounting for approximately 750 HNPCC kindreds worldwide6.
Lynch syndrome is caused by mutations in one of the mismatch repair (MMR) genes, including human mutL homolog 1 (hMLH1), human mutS homolog 2 (hMSH2), hMLH3, hMSH3, hMSH6, human PMS1 homolog 1, MMR system component (hPMS1), hPMS2, and human epithelial cell adhesion molecule7. Mutations in one of the alleles and additional somatic mutations result in defects of MMR system and accumulation of mutations in the genome, leading to carcinogenesis8. Pathogenicity of mutations associated with HNPCC is due to loss of main interaction domains or alterations in conformational structure of proteins encoded by MMR genes that disrupt their ability to interact other components of MMR pathway8.
Germline mutations of hMLH1 and hMSH2 genes account for greater than 90% of overall mutations9. hMLH1, the human homolog of bacterial MutL, is the main gene1. hMLH1 gene is located on 3p21 chromosome and contains 19 coding exons6. To date, more than 1600 variants of this highly polymorphic gene have been reported (http://genecards.org/cgi-bin/carddisp.pl?gene=MLH1&search=mlh1%23snp)10. The prevalence and type of mutations in these genes have not been similarly distributed in various populations that stem from genetic and geographical differences. Hence, identifying the common mutations in each region is potentially useful in the diagnosis of high-risk individuals, better management of disease, and designing effective prevention methods. The objective of this study was to investigate the germline mutations identified in hMLH1 gene in a population of HNPCC suspected families in Northwest of Iran. Moreover, the functional impact of the mutations was evaluated.
Materials and methods
Study population
In this paper, 30 patients with HNPCC of Azeri descent that met the Amsterdam II criteria (revised International Collaborative Group on HNPCC criteria 1998) were selected from the individuals recruited to the Ebne Sina Medical Genetic Center (Tabriz, Iran). According to Amsterdam II guideline, participants should have the following criteria; first, at least three relatives need to have cancer associated with HNPCC (colorectal, endometrial, stomach, ovary, ureter or renal-pelvis, brain, small bowel, hepatobiliary tract or skin [sebaceous tumors]). Second, one needs to be a first-degree relative of the other two. Third, at least two successive generations need to be affected. Fourth, at least, one of the relatives with CRC needs to have received the diagnosis before age 5011. Cancer diagnosis was confirmed by pathology reports. As a control group, 30 healthy volunteers were selected who had no cancer disorder either in themselves or their relatives. Personal and family cancer histories and written informed consent were obtained from all participants. The local ethical committee of Tabriz University of Medical Sciences approved this study.
Real-time polymerase chain reaction (RT-PCR) quantification
After taking 3 µl of peripheral blood sample of all subjects, RNA extraction was performed immediately. Total RNA of the peripheral blood was extracted using QIAamp RNA Blood Mini Kit (Cat No: 52304) according to the manufacturers instructions (Qiagen, Germany). About 500 ng of total RNA was used to synthesize the complementary DNA (cDNA), which was performed using Prime ScriptTM RT Reagent Kit (Cat No: RR037Q) according to manufacturers instructions (Takara, Japan).
Specific primers were designed using Primer 3 software12 to amplify the cDNA of hMLH1 gene (2484bp) in four overlapping fragments. Sequences of these particular primers are described in table 1. The primers were synthesized by Macrogen Company (Korea). RT-PCR was performed in a final volume of 25 µl reaction mixture, containing Red Mix: Taq DNA Pol 2X masters mix Red, 1.5 mM MgCl2 (No: 5200300-12121, Ampliqon, Denmark) which was diluted at a ratio of 1-1, 0.7 µl of each forward and reverse primers, and 1 µl of cDNA.
Table 1 List of primer sequences used for amplification of cDNA of hMLH1 gene and their respective sizes of PCR products
| Primer | Primer sequence (5→3) | Tm (°C) | Ta (C C) | Size (bp) |
|---|---|---|---|---|
| Forward 1 | CTTGGCTCTTCTGGCGCC | 63 | 63 | 675 |
| Reverse 1 | GGAGCGAATATTGTCCACGG | 60.5 | ||
| Forward 2 | ACAACATAGCCACGAGGAGAAAAG | 63.5 | 64 | 769 |
| Reverse 2 | ATTTTTGGCAGCCACTTCAGC | 59.4 | ||
| Forward 3 | GGAACAGAAGCTTGATGCATTTC | 61.1 | 64 | 638 |
| Reverse 3 | CATCTTCCTCTGTCCAGCCA | 60.5 | ||
| Forward 4 | CTGTGTGAATCCTCAGT | 50 | 68-58 | 822 |
| Reverse 4 | AAGGAATACTATCAGAAGGC | 54.3 |
Bp: base pair; Ta: annealing temperature; Tm: melting temperature.
cDNA: complementary DNA; PCR: polymerase chain reaction.
The reaction conditions were as follows: activation step at 95°C for 5 min, followed by 30 cycles of denaturation at 95°C for 40 s, specific annealing temperature for each fragment (as listed in Table 1) for 40 s, and 72°C for 45 s and a final extension at 72°C for 5 min. For the fourth fragment, the reaction conditions were as follows: activation step at 95°C for 3 min, followed by 25 cycles of denaturation at 95°C for 25 s, 68°C for 25 s, 72°C for 25 s, 25 cycles of denaturation at 95°C for 30 s, 58°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 7 min.
Germline mutation analysis
The PCR products were visualized by 2% agarose gel electrophoresis. The purified PCR fragments were sequenced by Macrogen Company (Korea). The reference sequence of hMLH1 gene was obtained from the NCBI database (http://www.ncbi.nlm.nih.gov/nuccore/NG.007109.1?form=4863&report=genbank), and INSIGHT-group database was used for mutation analysis (http://www.insight-group.org/mutations).
Functional analysis
Quantitative analysis was conducted using RT-PCR through the TaqMan Gene Expression Assays containing the FAM dye-labeled probes (TaqMan Pre-designed Gene Expression Products, Applied Biosystems, Foster City, CA, USA) and StepOne RT-PCR System (Applied Biosystems, Foster City, CA, USA). mRNA expression was conducted using the cDNA obtained through previous experiments. Moreover, the cDNA of 30 healthy controls was prepared and applied in the gene expression analysis. Each reaction mixture contained a total volume of 25 µl (master mix 12.5 µl, cDNA 4.5 µl, assay mix 3 µl, and H2O 5 µl). The quantitative RT-PCR conditions were as follow: 50°C for 1 min, 95°C for 5 min, then 40 cycles of 95°C for 30 s, and 60°C for 30 s, and finally 72°C for 30 s. The comparative CT method (2ΔΔCt method) was exerted to evaluate the expression as previously described by Livak and Schmittgen13. As the housekeeping gene, β-Actin mRNA transcript level was used for normalization of relative amounts of target mRNAs.
Statistical analysis
SPSS software version 21 (SPSS, Chicago, IL, USA) for data analysis was also used. The GraphPad Prism version 6.00 for Windows (GraphPad Software, Inc., San Diego, CA, USA, www.graphpad.com) was exerted to represent data by plots. Evaluation of the comparisons for mRNA expression level between groups was conducted through KruskalWallis nonparametric test. All data were shown as mean ± standard deviation with statistical significance set at 5%.
Results
In this study, 56.7% of patients were men with an average age of 47 ± 5.6 and 43.3% were women with an average age of 43.8 ± 6.8.
DNA sequencing results revealed four hMLH1 gene mutations in 50% of patients. The first variant (c.655A > G) was a polymorphism that occurred in exon 8 of hMLH1 gene that resulted in an A > G change at nucleotide position. This single nucleotide polymorphism (SNP) led to the substitution of the isoleucine amino acid by the valine at codon 219. The second variant (c.1959G > T) was a polymorphism which was caused by a change of G to T base in exon 17, which was found in only one of the patients.
In addition, two novel variants were found in the studied group that had not been reported before in any of the HNPCC mutation databases. The first variant was a transversion mutation (c.973C > A) which was detected in 4 out of 30 patients (13.33%). The second variant was a transversion mutation (c.1086C > A) which was found in exon 12 of six patients (20%). All of the detected variants were summarized in table 2 and figure 1.
Table 2 hMLH1 mutations and polymorphism detected by cDNA sequencing
| Gene | Exon region | Nucleotide change | Altered codon | Consequence | Number of affected patients | Type of variation |
|---|---|---|---|---|---|---|
| hMLH1 | Exon 8 | c. 655A > G | 219 | p.Ile219Val | 4 | Polymorphism |
| hMLH1 | Exon 11 | c. 973C > A | 325 | p.Arg325Arg | 4 | U.V. |
| hMLH1 | Exon 12 | c. 1086C > A | 362 | p.Ser362Ser | 6 | U.V. |
| hMLH1 | Exon 17 | c. 1959G > T | 653 | p.Leu653Leu | 1 | Polymorphism |
cDNA: complementary DNA; hMLH1: human mutL homolog 1.
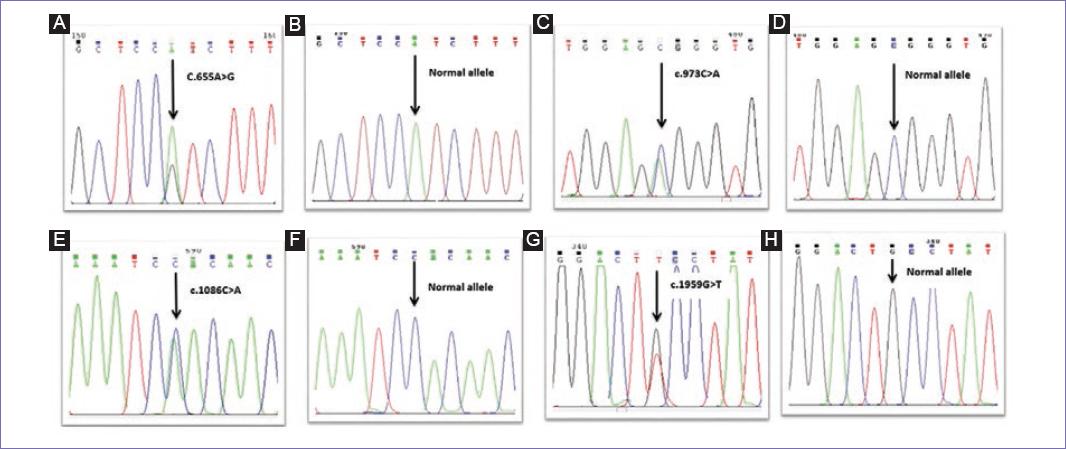
Figure 1 Sequence chromatograms of hMLH1 gene showing the germline mutation identified in the study group. A: c.655A > G polymorphism in exon 8; C: novel mutation, transversion mutation c.973C > A in exon 11; E: novel mutation, transversion mutation c.1086C > A in exon 12; G: c.1959G > T polymorphism in exon 17 (B, D, F, and H represent wild-type sequences).
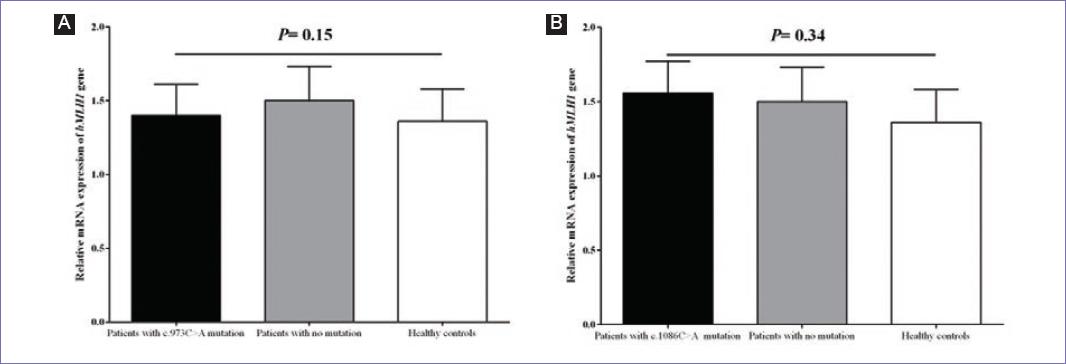
With respect to functional analysis (Fig. 2), it was found that mRNA expression level of hMLH1 had no statistically significant difference (p = 0.15) between patients who carried the novel c.973C > A mutation (n = 4) in comparison to the patients without any mutation (n = 4) as well as healthy controls (n = 30). Furthermore, no statistically significant difference (p = 0.34) was detected in the mRNA expression of hMLH1 gene in patients carrying the c.1086C > A novel mutation (n = 6) in comparison to the patients without any mutation (n = 4) as well as healthy controls (n = 30).
Discussion
CRC is one of the most common cancers in Iran. According to the recent epidemiological reports, the overall age-standardized incidence rates of CRC are higher in men (14.8/100,000 person-years) and the urban populations (35.4), relative to women (11.5) and the rural populations (17.1), respectively. The overall incidence rate of CRC has been reported to significantly increase from 2004 to 2013 in both men and women14. CRC accounts for 9.7% of all cancers incidences15, which is increasing continuously4. Furthermore, it shows a younger age distribution in Iranian population rather than western countries16. According to different molecular mechanisms, CRC is divided into two groups: sporadic (70% of all CRC cases) and inherited (nearly 20% of all CRC cases). HNPCC or Lynch syndrome is the most common form of hereditary CRC, which constitutes 5-8% of all CRC cases17.
The defects of different genes of MMR system result in HNPCC syndrome. Among these genes, hMLH1 gene, which is the most frequently mutated gene, plays the main role in the etiopathogenesis of this syndrome18. Here, in this study, the hMLH1 gene germline mutations in 30 HNPCC patients were investigated using RT-PCR and cDNA sequencing methods. cDNA sequencing is a useful technique in recognizing the mutations and decreases the cost of sequencing mainly19. We found four variants in the studied population. The first variant was c.655A > G in a conserved region of exon 8 of 4 patients. This SNP leads to the substitution of the isoleucine by the valine at position p.219 in the hMLH1 protein, but the influence of this common missense polymorphism on colon cancer risk is poorly understood4. There are conflicts on the importance of this variant in the hMLH1 protein performance. On the one hand, both mutant and wild alleles result in non-polar and pH-neutral amino acid residues20. On the other hand, functional analyses suggest that this variant may impress the efficiency of DNA repair system21. In another study, it was found that in male patients, G-allele carriers (AG or GG genotype) had higher risk to develop CRC relative to homozygous AA wild-type individuals. Therefore, sexuality should be considered in analyzing the results. Furthermore, patients with G-allele show a better pattern of the disease, angiogenesis, and metastasis, and disease recurrence are less observed among these people22. A few reports have shown that the hMLH1 p.Ile219Val polymorphism is not entirely benign and is associated with nearly 5-fold increased risk of ulcerative colitis, which is a major risk factor for colon or rectum cancer23,24 and is related to early onset of CRC in patients25. Decreasing in hMLH1 protein expression is associated with this polymorphism among sporadic CRC cases from Korea26. Thus, further extensive studies are necessary to clarify the real impact and importance of this polymorphism toward the susceptibility to CRC.
The second variant detected in our investigations was c.1959G > T (p.Leu653Leu) in exon 17 of hMLH1 gene in only one of the patients. This variation has been reported as a neutral variant that does not have a consequential effect on MMR system. According to available information from the insight group database, it is suggested that this mutation forms alternative splicing site, which causes exon skipping, and since there is no phenotypic consequence, it cannot be considered as a rare polymorphism27.
On the other hand, two novel variants were recognized in this study. The novelties of these variants are based on the insight database (www.INSiGHT-group.org). The c.973C > A variant is located in the middle part of exon 11 of hMLH1 gene. Since exon 11 encodes the terminal part of MutS interaction domain, hence this variant can affect the hMLH1 interaction with hMSH2 or hMSH6, which are necessary for accurate performance of MMR system. The second novel variant was c.1086C > A in the primary part of exon 12 of hMLH1 gene. Since this region has no interaction with other MMR proteins, thereupon, it seems that this variant has no important effect on hMLH1 gene performance. However, the high frequencies of these variations indicate the predominance of these variants among Iranian population. On the other side, functional analysis indicated that these novel mutations did not impress the mRNA level of hMLH1 gene. It can be speculated that other regulatory feedback mechanisms might correct the mRNA expression and final functional impression of these mutations.
Considering all the results, this study identified two already reported polymorphisms (including c.655A > G and c.1959G > T) as well as two novel variants (c.973C > A and c.1086C > A) in hMLH1 gene for the 1st time in HNPCC patients. The novel mutations had no impression of the mRNA expression level of hMLH1 gene. The most reliable way to diagnose Lynch syndrome is to detect a mutation in the MMR genes in the suspected patients. By investigating and determining the common mutations in each region, researchers will be able to use genetic testing for early detection of CRC in the relatives of the patients. This will result in decreasing the mortality and morbidity of CRC and better management of the disease.

