











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La obesidad es la clave
La obesidad ya ha sido reconocida por la Organización Mundial de la Salud (OMS) como un padecimiento epidémico mundial que afecta tanto a países desarrollados como en vía de desarrollo (World Health Organization, 2022). En México ya se ha declarado una emergencia sanitaria debido al incremento de personas con obesidad y diabetes, por lo que la obesidad es considerada actualmente como un problema de salud pública (Secretaría de Salud, Instituto Nacional de Salud Pública, & Instituto Nacional de Estadística y Geografía, 2019). En la actualidad está ampliamente demostrado que la sobrealimentación hipercalórica da como resultado inflamación metabólica, una forma de inflamación crónica de bajo grado y “estéril”, es decir, en ausencia de cualquier infección microbiana sistémica o local demostrable (Hotamisligil, 2006; Schenk, Saberi, & Olefsky, 2008). Esta activación de vías inflamatorias inducida por sobrealimentación hipercalórica se ha observado en una variedad de tejidos periféricos que se comunican regularmente con los centros energéticos, incluidos los tejidos adiposo blanco (Villarroya, Cereijo, Gavaldà-Navarro, Villarroya, & Giralt, 2018), hígado (van der Heijden et al., 2015), músculo esquelético (Wu & Ballantyne, 2020), entre otros. Actualmente se ha demostrado una estrecha relación entre la inflamación de tejidos periféricos inducida por la obesidad con la reactividad de las células de la glía en el sistema nervioso central (SNC), principalmente en el hipotálamo (Guillemot-Legris & Muccioli, 2017), lo que forja un antecedente como posible responsable de las modificaciones funcionales del hipotálamo que regulan los mecanismos de hambre y saciedad. Con base a lo anterior, esta revisión se enfoca en la descripción general de la disfunción metabólica asociada a la obesidad y su participación en la alteración de la regulación hipotalámica, provocando neuroinflamación y modificaciones en la conducta alimentaria. En la tabla siguiente se presentan las abreviaturas utilizadas en este artículo (Tabla 1).
Tabla 1 Abreviaturas de términos utilizadas en este artículo.
| A1: Estado de activación de los astrocitos similar a M1 | EP2: Receptor a Prostaglandina E2 | NF-kB: Factor Nuclear Kappa B |
| A2: Estado de activación de los astrocitos similar a M2 | GABA: ácido gamma amino butírico | NPY: Neuronas que expresan Neuropéptido Y |
| AGL: Ácidos Grasos Libres | GFAP: Proteína Ácida Fibrilar Glial | OMS: Organización Mundial de la Salud. |
| AgRP: Péptido Relacionado a Agouti | GM-CSF: Factor Estimulante de Colonias de Granulocitos y Macrófagos. | PAMPs: Patrones Moleculares Asociados a Patógenos |
| AGS: Ácidos Grasos Saturados | IFN-γ: Interferon γ. | POMC: Neuronas que expresan propiomelanocortina |
| ARC: Núcleo Arqueado del Hipotálamo | IkB: Inhibidor del Factor Nucelar Kappa B | PPAR: receptores activados por proliferadores peroxisomales |
| ARNm: Ácido Ribonucléico Mensajero | IL: Interleucina. | PRR: Receptores de Reconocimiento de Patrones |
| BDNF: Factor Neurotrófico Derivado de Cerebro | iNOS: Enzima Óxido Nítrico Sintasa Inducible | ROS: Especies Reactivas de Oxígeno |
| BHE: Barrera Hemato Encefálica | JNK: Cinasa c-Jun N-terminal | SNC: Sistema Nervioso Central. |
| CART: Neuronas de transcripto regulado por cocaína y anfetamina | LPS: Lipopolisacárido. | TGF-β: Factor de Crecimiento Transformante β |
| CD: Cluster de diferenciación | M0: Macrófago o microglía quiescente. | TH1: Perfil de activación tipo 1 de linfocitos cooperadores. |
| CCR2: Receptor de la quimicoina MCP-1 o CCL2 | M1: Macrófago o microglía proinflamatoria. | TH2: Perfil de activación tipo 2 de linfocitos cooperadores. |
| COX-1: Ciclooxigenasa 1 | M2: Macrófago o microglía antiinflamatoria. | TLR: Receptor tipo Toll |
| CX3CL1: Quimiocina Fractalquina. | MC4R-KO: Ratones deficientes en el receptor de melanocortina | TNFɑ: Factor de Necrosis Tumoral ɑ. |
| DAG: Dieta Alta en Grasa | MCP-1: Proteína quimioatrayente de monocitos 1, también conocida como CCL2. | |
| DAMPs: Patrones Moleculares Asociados a Daño Celular | MHC-I: Molécula del complejo principal de histocompatibilidad tipo I | |
| EM: Eminencia Media | MTA: Macrófagos de Tejido Adiposo |
La obesidad como punto de partida de la neuroinflamación hipotalámica
La obesidad, lejos de estar relacionada con la ganancia de peso, generalmente se asocia con un cúmulo de desórdenes conocidos como síndrome metabólico, cuya etiología consta principalmente de la interacción entre predisposiciones genéticas y factores ambientales que resultan en una inflamación sistémica de bajo grado que afecta a numerosos tejidos incluidos el hígado, el tejido adiposo y el SNC (Lumeng & Saltiel, 2011), y es la base para el desarrollo y progreso de diferentes condiciones patológicas asociadas con la obesidad y la resistencia a la insulina. La acumulación crónica y excesiva de energía en el tejido adiposo provoca en el individuo sobrepeso y obesidad.
Los adipocitos son las células del tejido adiposo que producen y secretan un gran número de proteínas que se encargan de regular el metabolismo, el consumo energético y el almacén de grasa, estos factores se conocen como adipocinas y cumplen diferentes funciones (Cuspidi, Tadic, & Grassi, 2014). Algunas proteínas como la adiponectina o la leptina tienen funciones citoprotectores y reguladoras, pero otras como las citocinas proinflamatorias como IL-1β, IL-18, TNF-α, MCP-1, entre otras, contribuyen a la desregulación de la homeostasis de la glucosa, regulación de la ingesta alimentaria y de respuestas inflamatorias (Wang & He, 2018; Holland et al., 2011). Por su parte, la disminución de la secreción de algunas adipocinas como la adiponectina o la neuregulina 4, así como la sobreregulación de otras adipocinas como la calprotectina e IL-6 favorecen en conjunto la resistencia a la insulina y la tolerancia a la glucosa (Kawai et al., 2021; Czech, 2020; Holland et al., 2011). La obesidad induce una compleja remodelación del tejido adiposo, que se expande para adaptarse a la ingesta calórica excesiva y cambia notablemente su estructura y composición celular. Esta expansión está mediada por un aumento del tamaño de los adipocitos (hipertrofia) y en el número de adipocitos (hiperplasia) (Stenkula & Erlanson-Albertsson, 2018). La hipertrofia es un proceso celular altamente prevalente durante la obesidad, sin embargo, la hiperplasia además de ser un mecanismo importante en la obesidad, también es un proceso fisiológico que está presente en sujetos sanos, es la alteración en la tasa de recambio de los adipocitos lo que distingue a la obesidad de la homeostasis, aunque sus mecanismos no han sido claramente identificados (Sun, Kusminski, & Scherer, 2011).
En estudios realizados en humanos no obesos, se reportó que la vida media de las células en el tejido adiposo es de aproximadamente 8.3 años, lo que implica una lenta tasa de recambio, mientras que en adipocitos de personas con obesidad existe un desbalance en el índice conocido como radio apoptótico-adipogénico, en el cual hay un incremento de adipogénesis respecto a la cantidad de adipocitos eliminados por apoptosis (Spalding et al., 2008). Esta observación sugiere que un mecanismo responsable del aumento en el tejido adiposo puede ser por el reclutamiento de preadipocitos a través de factores quimiotácticos secretados por los adipocitos hipertróficos existentes en el tejido adiposo de los pacientes obesos (Marques, Hausman, & Martin, 1998); sin embargo, los mecanismos específicos y las moléculas implicadas aún no se han descrito por completo.
La hipertrofia e hiperplasia de los adipocitos también promueve la infiltración de células inmunitarias, predominantemente macrófagos derivados de monocitos que son reclutados por la acción de la quimiocina MCP-1. Esta quimiocina, también conocida como CCL2, es secretada por los adipocitos y regula el microambiente inflamatorio del tejido adiposo promoviendo la quimioatracción de monocitos-macrófagos de la periferia al estimular los receptores CCR2, cuya consecuencia es reclutar estas células para sinergizar con la respuesta proinflamatoria en el tejido adiposo. Durante la obesidad se sobrerregula la producción tanto del ligando como del receptor (Reilly & Saltiel, 2017; Panee, 2012). Los macrófagos son las células inmunitarias dominantes en el tejido adiposo y muestran una gran heterogeneidad en sus funciones que reflejan sus microambientes metabólicos e inmunitarios locales. Los macrófagos generalmente existen en dos poblaciones distintas: 1) macrófagos activados de la forma clásica o M1, éstos son proinflamatorios y polarizados por lipopolisacárido (LPS), un glicolípido que forma parte de los principales componentes de la membrana externa de las bacterias Gram-negativas y que es descrito como un potente activador de la respuesta inflamatoria (Fujihara et al., 2003), ya sea solo o en asociación con citocinas TH1 como IFN-γ, GM-CSF, y producen pro-citocinas inflamatorias tales como interleucina-1β (IL-1β), IL-6, IL-12, IL-23 y TNF-α; y 2) los macrófagos activados de forma alternativa o M2, antiinflamatorios e inmunorreguladores polarizados por citocinas TH2 como IL-4 e IL-13 y productores de citocinas antiinflamatorias como IL-10 y TGF-β. Los macrófagos M1 y M2 tienen diferentes funciones y perfiles transcripcionales (Shapouri-Moghaddam et al., 2018).
Las interacciones entre macrófagos y adipocitos influyen tanto en el metabolismo como en la inflamación, por lo que es necesario un delicado equilibrio entre las poblaciones polarizadas de macrófagos para mantener una función adecuada de los adipocitos. Las señales inflamatorias en el tejido adiposo de las personas con obesidad activan el cambio fenotípico de los macrófagos de M1 a M2 (Sun et al., 2011). El depósito excesivo de grasa ectópica en la obesidad provoca trastornos de la homeostasis energética e inflamación crónica de bajo grado en los tejidos metabólicos. En particular, el reclutamiento y la activación de los macrófagos del tejido adiposo inducidos por la obesidad juegan un papel clave en la patogénesis. En un artículo histórico de 2003, se informó que los ratones y los humanos con obesidad tenían un aumento de macrófagos del tejido adiposo (MTA), lo que se correlacionaba con su grado de adiposidad. Esto sugirió que el tejido adiposo puede reclutar macrófagos adicionales en respuesta a estímulos (Weisberg et al., 2003). En estados de no obesidad, sensibles a la insulina, los MTA se polarizan hacia un estado M2 con expresión de IL-10 y arginasa 1. Al principio del tratamiento con una dieta alta en grasa (DAG), los adipocitos se hipertrofian y liberan quimiocinas que inducen el reclutamiento de MTA polarizados en M1 con baja expresión de IL-10 y una mayor producción de iNOS y TNF-α (Lumeng, Bodzin, & Saltiel, 2007). En estas primeras etapas de obesidad leve con sensibilidad a la insulina, los MTA residentes polarizados M2 pueden proteger parcialmente a los adipocitos de estos factores inflamatorios y pueden bloquear la polarización M1, con la expresión de citocinas antiinflamatorias como IL-10 y TGF-β o marcadores como la arginasa 1 (Mills, 2012;Lumeng, Bodzin, & Saltiel, 2007). Con el aumento de la adiposidad, los MTA CCR2+ reclutados forman estructuras en forma de corona y superan los efectos protectores de los macrófagos M2, lo que estimula a tener un ambiente proinflamatorio dominado por TNF-α e iNOS. Estos factores generan resistencia a la insulina en los adipocitos, activan las vías de JNK y NF-κB, alteran la secreción de adipocinas y conducen a niveles circulantes excesivos de ácidos grasos libres (AGL) debido a la lipólisis de los adipocitos y al deterioro de la lipogénesis (Lumeng, Bodzin, & Saltiel, 2007).
Con el aumento de MTA proinflamatorios y el reclutamiento de otros leucocitos como linfocitos TH1 y T citotóxicos, se incrementa la producción de citocinas proinflamatorias como TNFɑ, IL-1β e IL-6, las cuales además de promover acciones dentro del tejido adiposo, migran a través de la circulación sanguínea, contribuyendo al desarrollo a largo plazo de alteraciones metabólicas como la resistencia a la insulina, síndrome metabólico e inflamación crónica de bajo grado a nivel sistémico, entre otras (Kawai et al., 2021; Wu & Ballantyne, 2020). La citocina TNFɑ, fue la primera molécula descrita como el enlace entre la inflamación local y la inflamación sistémica en la obesidad, evolucionando a un concepto conocido como “inflamación metabólica”, ya que se secreta principalmente por los MTA y en menor proporción por los adipocitos, fibroblastos, neutrófilos y linfocitos TH1; además es capaz de activar la señalización intracelular por las vías de MAPK y NF-kB en conjunto con la IL-6 e IL-1β (Artemniak-Wojtowicz, Kucharska, & Pyrżak, 2020). La IL-6 además de tener propiedades como citocina pro-inflamatoria, se ha descrito que estimula a los hepatocitos para producir y secretar proteína C reactiva, marcador metabólico de inflamación, además de inhibir la adiponectina (Sopasakis et al., 2004; Wärnberg, Moreno, Mesana, Marcos, & AVENA group, 2004).
La inflamación crónica de bajo grado que genera la expansión atípica del tejido adiposo derivada de la obesidad contribuye al desarrollo de inflamación en otros órganos y los afecta a largo plazo, por ejemplo promoviendo hígado graso, enfermedades cardiovasculares, diabetes tipo 2, asma, algunos tipos de cáncer, e incluso neuropatologías inflamatorias y neurodegenerativas (Artemniak-Wojtowicz, Kucharska, & Pyrżak, 2020).
Si bien los primeros órganos afectados durante la inflamación sistémica son aquellos asociados al metabolismo de ácidos grasos y carbohidratos, también se afectan otros órganos y sistemas como el SNC, por la exposición continua a AGL y neurotoxinas como el LPS (Moreno-Navarrete et al., 2017).
Por otro lado, la peroxidación lipídica y la hidrólisis de AGL en el hígado y el endotelio vascular en pacientes con obesidad, promueven el transporte de AGL a tejidos periféricos. Esto genera una mayor concentración de adipocinas y de varias hormonas que activan la vía proinflamatoria que es la base de la patogénesis de la arteriosclerosis y sus complicaciones crónicas como el infarto al miocardio y accidente cerebrovascular, entre otras. En conjunto con la inflamación sistémica y el estrés oxidante, los AGL promueven la permeabilidad de diversas barreras celulares como la barrera hematoencefálica (BHE), cuya función es la internalización selectiva de nutrientes y señales periféricas (Stranahan, Hao, Dey, Yu, & Baban, 2016). El aumento de permeabilidad en esta permite la entrada de factores proinflamatorios y otros estímulos periféricos como el LPS y leucocitos de la periferia que migran hacia el SNC en respuesta a la neuroinflamación (Kim, Glendining, Grattan, & Jasoni, 2016; Stranahan, Hao, Dey, Yu, & Baban, 2016).
Estos estímulos desencadenan respuestas a través de la estimulación de los receptores de reconocimiento de patrones (PRR), actuando como patrones moleculares asociados a patógenos (PAMPs). Como ya se mencionó previamente, el LPS es un potente activador de la respuesta inmunitaria proinflamatoria, en específico de la desregulación del microbiota intestinal derivada de los cambios dietéticos. Este desbalance en la microbiota promueve el incremento de los niveles circulantes de LPS, lo cual desencadena la activación inmunitaria y glial (Reilly & Saltiel, 2017). Otro ejemplo son las alarminas o patrones moleculares asociados a daño celular (DAMPs) como los AGL o el colesterol que también estimulan respuestas similares tanto en las células inmunitarias periféricas como en la glía (Kim et al., 2016). Se ha descrito que la estimulación por estas moléculas está dirigida a través de la señalización de los receptores tipo-Toll 2 y 4 (TLR2 y TLR4), que poseen tanto las células inmunitarias (neutrófilos, macrófagos, células dendríticas, etc.) como algunas células epiteliales, endoteliales y adiposas de diversos tejidos que desencadenan la vía de NF-kB, promoviendo la activación inflamatoria para la posterior expresión de factores proinflamatorios, adipocinas, factores de complemento y factores de crecimiento en la periferia (Nishimura, Manabe, & Nagai, 2009; Stolarczyk, 2017).
A nivel celular, la exposición de las neuronas a una dieta hipercalórica abundante en lípidos como la DAG induce mecanismos adaptativos como la autofagia y el estrés del retículo endoplásmico que promueven el daño neuronal a largo plazo, sin embargo no son las únicas células residentes del parénquima cerebral que resultan afectadas por la desregulación metabólica e inflamación sistémica asociada a la dieta. Más del 50% de la composición celular del cerebro es no neuronal, incluidos los componentes gliales, vasculares y periventriculares (Colonna & Butovsky, 2017). Los astrocitos y la microglía son los más abundantes de esos tipos de células especializadas que, además de comprender gran parte de la arquitectura básica del parénquima cerebral, mantienen la BHE, apoyan el metabolismo neuronal, protegen y reaccionan a la lesión tisular local. Estas células tienen una gran plasticidad para combatir infecciones, eliminar neuronas dañadas y dirigir el proceso de restauración (Colonna & Butovsky, 2017; Liu, Liu, Bao, Bai, & Wang, 2020), por lo cual se les considera no solo como células de soporte metabólico neuronal, sino también como células sensibles a las señales tanto metabólicas como inflamatorias de origen periférico, adaptándose y respondiendo a los estímulos de acuerdo con el tipo de estímulo, con capacidad de contribuir al ambiente inflamatorio secretando citocinas y quimiocinas (Dorfman & Thaler, 2015). El ambiente proinflamatorio derivado de la inflamación sistémica crónica de bajo grado impacta de igual forma en la glía, debido al aumento en la permeabilidad de la BHE que permite la difusión libre de las citocinas proinflamatorias y LPS de la circulación periférica, polarizando la microglia a un perfil proinflamatorios. Esto se observa principalmente en el núcleo arqueado del hipotálamo (ARC) que es el centro neuronal especializado que integra las señales hormonales y nutricionales captadas desde la circulación periférica para regular la conducta alimentaria, la homeostasis energética y el metabolismo de glucosa (Kim et al., 2016; Waterson & Horvath, 2015; Morton, Meek, & Schwartz, 2014).
En el SNC, la respuesta inmunitaria se encuentra estrictamente controlada y regulada por mecanismos específicos que permiten responder a estímulos pro-inflamatorios agudos, reduciendo el potencial daño neurotóxico (Jurga, Paleczna, & Kuter, 2020). La microglía es la principal célula glial encargada de regular la inmunidad en el SNC y es considerada como el macrófago residente del SNC (Uriarte Huarte, Richart, Mittelbronn, & Michelucci, 2021).
Participación de la glía hipotalámica en la regulación energética
La alimentación DAG y la acumulación de AGL también se asocian con la acumulación de células gliales activadas, incluidas la microglía M1 y los astrocitos reactivos con perfiles proinflamatorios, similar a la infiltración tisular asociada a la obesidad en tejidos periféricos por células inmunes activadas, principalmente macrófagos y otras clases de leucocitos (De Souza et al., 2005; Milanski et al., 2009; Posey et al., 2009). Las células gliales se dividen principalmente en astrocitos, oligodendrocitos, células ependimarias y microglía, que componen, junto con las células endoteliales, la población no neuronal del parénquima cerebral (Jessen, 2004). La glía representa una población diversa de células no neuronales en el sistema nervioso periférico, central o autónomo. Son células dinámicas que participan en el metabolismo cerebral, la comunicación entre neuronas y regulan la respuesta inmune (Xu, Lu, Shao, Zhang, & Zhang, 2020). La glía juega un papel crítico en la actividad del SNC durante el desarrollo, la salud y la enfermedad (Jäkel & Dimou, 2017). Varios estudios han demostrado que las células gliales que se encuentran en el hipotálamo son esenciales para regular el metabolismo energético tanto en condiciones fisiológicas como patológicas (Freire-Regatillo, Argente-Arizón, Argente, García-Segura, & Chowen, 2017). Estas células, principalmente los astrocitos, detectan cambios en la disponibilidad de nutrientes a través de la vía de la leptina y sus receptores (hormona reguladora del apetito), lo que permite que en conjunto con las neuronas del ARC controlen el apetito y el consumo energético (Yang, Qi, & Yang, 2015). La ingesta excesiva de nutrientes y/o la disminución del gasto energético pone en riesgo el mantenimiento a largo plazo de la homeostasis energética. En los siguientes apartados, se discutirá el impacto de los cambios dietarios como la DAG sobre la función de los astrocitos y la microglía.
La microglía son células gliales que se han identificado como uno de los reguladores de la homeostasis energética en el parénquima cerebral (Yang, Qi, & Yang, 2015). Estas células representan aproximadamente el 15% de las células totales en el SNC y el 32% de células de la glía (Lawson, Perry, & Gordon, 1992). Las células de la microglía son macrófagos residentes del SNC que se establecen en el parénquima cerebral desde las etapas tempranas del neurodesarrollo a partir del saco vitelino (Yang et al., 2015). La microglía actúa como célula de primera respuesta a una variedad de alteraciones en el SNC, incluidos patógenos, isquemia y lesiones neuronales. Estas células cambian su morfología, de ser altamente ramificadas (tipo-R) a tomar una morfología ameboide (tipo-HR). Cuando se encuentran en un estado quiescente (homeostático, M0 o de vigilancia inmunitaria) permanecen con una morfología tipo-R, sin embargo, cuando se activan pasan a tomar la morfología tipo-HR y son reclutadas rápidamente a las regiones de daño o muerte celular donde proliferan in situ. (Ransohoff & Cardona, 2010). De acuerdo con el estímulo que detecta, la microglía ameboide adapta su perfil de expresión y sus funciones celulares a dos diferentes estadios celulares nombrados en homología a la nomenclatura de los macrófagos periféricos: M1, para un perfil proinflamatorio con secreción de citocinas como TNF-α, IL-1β, IL-6, IL-12, entre otras; y M2, que se caracteriza por tener un perfil anti-inflamatorio y reparador de tejido, con predominancia de secreción de citocinas como IL-10, TGF-β, entre otros (Dorfman & Thaler, 2015). La microglía responde a la presencia de PAMPs y DAMPs de forma similar a tejidos fuera del cerebro (Kigerl et al., 2014). La inyección sistémica de una única dosis de LPS promueve cambios en la morfología de la microglía hipotalámica similares a los generados durante su activación tipo M1. Estos cambios se observan 8h después de la inyección, alcanzando su punto máximo después de 24h y su recuperación después de 7 días (Buttini, Limonta, & Boddeke, 1996). Esto va acompañado del aumento en la expresión de MHC-I y citocinas inflamatorias (TNF-α, IL-1ß e IL-6), y también en la activación de COX-1 y NF-κB (García-Bueno, Serrats, & Sawchenko, 2009). En estadio M1, la microglía realiza funciones fagocíticas y produce mediadores proinflamatorios incluyendo citocinas, quimiocinas, especies reactivas de oxígeno (ROS) y óxido nítrico que en conjunto neutralizan al elemento agresor y mantienen un medio neuroprotector. Sin embargo, su activación prolongada puede resultar en inflamación crónica que contribuye a la progresión de enfermedades neurodegenerativas y neoplásicas (Glass, Saijo, Winner, Marchetto, & Gage, 2010). Por otro lado, aproximadamente el 60% de la población glial en el parénquima cerebral está compuesto de astrocitos, una célula derivada de la cresta neural que provee de soporte trófico, sináptico y metabólico a las neuronas y participa activamente en la respuesta neuroinflamatoria, entrando en el estadio de astrocitos reactivos (Vasile, Dossi, & Rouach, 2017). De forma homeostática, los astrocitos hipotalámicos han sido objeto de estudio en el campo de la neuroendocrinología, por su función reguladora del metabolismo junto con las neuronas del ARC, inhibiendo la actividad de las neuronas AgRP (orexigénicas) a través de los receptores A1 para adenosina pre- y post- sinápticos, obteniendo una función anorexigénica indirecta (Yang et al., 2015) y por medio de la señalización asociada a la vía de insulina y detección de viabilidad de nutrientes en el parénquima cerebral (García-Cáceres et al., 2016). Contrario a estos datos, en 2021 Varela y col. estudiaron la activación de neuronas AgRP dirigida por ghrelina, promoviendo la liberación de GABA que llevó a los astrocitos a incrementar su cobertura con sus pies astrocíticos a dichas neuronas, desencadenando liberación de prostaglandina E2 e incrementando su excitabilidad a través de los receptores EP2, fungiendo como orexigénicos (Varela et al., 2021), lo que sugiere la formación de un circuito metabólico funcional dinámico de los astrocitos con las neuronas hipotalámicas.
Neuroinflamación hipotalámica asociada a dietas hipercalóricas.
La inflamación hipotalámica asociada a la obesidad se ha observado en el estudio con ratas alimentadas durante 16 semanas con DAG (De Souza et al., 2005). Este trabajo mostró que el consumo de una dieta rica en grasa como la DAG induce la expresión de las citocinas proinflamatorias: TNF-α, IL-1β e IL-6. En otros estudios, se demostró que la expresión de estos mismos genes inflamatorios en el hipotálamo fueron inducidos rápidamente desde las primeras 24 h después del inicio de la DAG en ratones y ratas. Además de la presencia de estas citocinas, el incremento de lípidos asociados con la dieta, especialmente esfingolípidos como la ceramida, induce la resistencia a la insulina dependiente de TLR4 (Holland et al., 2011; Thaler et al., 2012; Zhang, Reichel, Han, Zuniga-Hertz, & Cai, 2017). Una de las distinciones importantes entre la inflamación metabólica inducida por la dieta en los tejidos periféricos y la que ocurre en el hipotálamo se relaciona con la latencia. Los estudios muestran que la inflamación en respuesta a la alimentación se desarrolla más rápidamente en el hipotálamo en comparación con los tejidos periféricos. Por lo tanto, la inflamación hipotalámica inducida por una dieta hipercalórica precede a la aparición de obesidad y ocurre mucho antes que la inflamación o las alteraciones metabólicas en los tejidos periféricos (Guillemot-Legris & Muccioli, 2017).
En el hipotálamo, el ARC es el principal participante en la regulación del hambre y la saciedad, en este núcleo se ubican las dos poblaciones neuronales más importantes y que tienen funciones antagonistas, conocidas como las neuronas orexigénicas que secretan péptido relacionado a Agouti (AgRP) y neuropéptido Y (NPY), y las neuronas anorexigénicas que secretan proopiomelanocortina (POMC) y son reguladas transcripcionalmente por cocaína y anfetamina (CART). La comunicación entre estas neuronas es a través de señales periféricas provenientes del tejido adiposo y el páncreas, ya que tanto la insulina como la leptina llegan al hipotálamo por distribución sanguínea y ejercen acción sobre las neuronas AgRP/NPY y POMC/CART, regulando la conducta alimentaria (Morton et al., 2014; Waterson & Horvath, 2015).
Estudios en tejidos periféricos tales como el hígado y el tejido adiposo mostraron que el consumo excesivo de lípidos y carbohidratos determina el establecimiento de un proceso inflamatorio crónico de intensidad leve, el cual interfiere con la señalización de la insulina y lleva a un estado de intolerancia a la glucosa (Hotamisligil, 2006). Las grasas y los hidratos de carbono participan en la neuroinflamación inducida por una dieta hipercalórica. Los primeros estudios analizaron el efecto de las dietas que contienen grasas saturadas y cuyo consumo promueve obesidad y neuroinflamación (Moreno-Navarrete et al., 2017; Vessby, 2003). Posteriormente se demostró que las dietas con una alta carga glucémica pueden afectar negativamente a los marcadores inflamatorios de riesgo (aumento de leptina, proteína C reactiva, entre otros) en individuos con obesidad sometidos a dietas ricas en hidratos de carbono (Dhingra et al., 2007;Kasim-Karakas, Tsodikov, Singh, & Jialal, 2006).
Las dietas altas en lípidos y proteínas promueven cambios en los órganos periféricos con el desarrollo de hiperplasia, hipertrofia e hipoxia de los adipocitos, incremento de producción de radicales libres y el desarrollo de estrés oxidante, inhibición de las vías de receptores activados por proliferadores peroxisomales (PPAR) y estrés de retículo endoplásmico en diversas estirpes celulares que van desde los mismos adipocitos, hasta músculo y células inmunitarias (Kawai et al., 2021; Artemniak-Wojtowicz, Kucharska, & Pyrżak, 2020), lo que impacta directamente en la activación de vías proinflamatorias como vía de los inflamasomas, activación de TLR y NF-kB, así como de la producción de citocinas y quimiocinas proinflamatorias que cruzan la BHE, llegan al hipotálamo y promueven el desarrollo de neuroinflamación, alterando la comunicación celular en esa área cerebral, específicamente en el ARC, con el desarrollo de gliosis (Seong, Kang, Sun, & Kim, 2019).
El descubrimiento del incremento de citocinas proinflamatorias como TNFɑ, IL-1β, e IL-6 en el ARC y el hipotálamo lateral de ratones, en respuesta a la alimentación DAG, permitió sugerir la relación entre la obesidad y la inflamación del hipotálamo en la homeostasis energética (De Souza et al., 2005). Este proceso de inflamación desencadena la activación de cinasas, como c-Jun e IkB, que inhiben los mediadores que están corriente abajo de las vías de la insulina y la leptina, generando resistencia a insulina/leptina en algunas neuronas hipotalámicas (Zhang et al., 2017; Milanski et al., 2009; Posey et al., 2009; Shoelson, Lee, & Goldfine, 2006) . La inflamación hipotalámica se identificó principalmente en el ARC y el consumo crónico de dietas ricas en lípidos lleva a la pérdida de las neuronas POMC, encargadas de las señales anorexigénicas en el circuito del consumo de alimento (Seong, Kang, Sun, & Kim, 2019; Thaler et al., 2012). Esta modificación de la señalización por POMC y el mantenimiento de la señalización de las neuronas orexigénicas NPY/AgRP, en conjunto con la gliosis de tanto astrocitos como microglia y la promoción de respuestas proinflamatorias in situ, son las que promueven que el sujeto en condiciones de obesidad continue buscando el consumo de alimento, especialmente aquella rica en lípidos y carbohidratos (Seong, Kang, Sun, & Kim, 2019).
Microglía y la alteración en la homeostasis energética
Diferentes estudios con ratones han demostrado que el ARC es sensible a los ácidos grasos saturados (AGS) (ácidos láurico, palmítico y esteárico) (Baufeld, Osterloh, Prokop, Miller, & Heppner, 2016; Valdearcos et al., 2014). En el hipotálamo de los ratones el consumo de AGS promueve una respuesta microglial rápida, induce gliosis (proliferación e hipertrofia de varios tipos de células gliales, incluidos astrocitos reactivos, microglía M1 y oligodendrocitos activados), cambios rápidos en la morfología de la microglía e inflamación hipotalámica (Baufeld et al., 2016). Inicialmente, estos cambios morfológicos están restringidos a la región del ARC cerca de la transición con la eminencia media (EM), y se acompañan de una mayor expresión de citocinas inflamatorias como IL-6, IL-1β y TNFα y otros marcadores de inflamación, tales como CD74 e IRF8. La respuesta inflamatoria del hipotálamo a los DAG ocurre en un modo bifásico. Durante la primera semana de una DAG, los niveles de ARNm y de proteína de las citocinas hipotalámicas, como TNF-α, IL-1β e IL-6, así como de las quimiocinas, como MCP-1 y CX3CL1, se incrementan (Morari et al., 2014), pero en estudios de seguimiento temporal, se ha demostrado que existe un periodo de regulación del estado neuroinflamatorio, el cual vuelve a incrementarse con el consumo de la DAG de forma crónica. En un estudio del curso temporal del cambio dietario a una DAG en ratas Long Evans y en ratones C57BL6/J, se observa cómo se desarrolla en el hipotálamo una primera respuesta proinflamatoria ante la DAG dentro de los primeros tres días, con incremento de marcadores como IL-6, IL-1β y mediadores de la vía de NF-kB. Sin embargo, los niveles de estos marcadores disminuyen al día siete. Con la persistencia del consumo de la DAG, después de 4 semanas se incrementan nuevamente los marcadores inflamatorios y se mantienen hasta el día 28, último tiempo de estudio (Thaler et al., 2012). La naturaleza bifásica de la respuesta inflamatoria en el hipotálamo en modelos experimentales de obesidad ocurre de manera similar a la respuesta inflamatoria bifásica clásica en otros tejidos. Una de las razones de este comportamiento bifásico asociado a la obesidad es que la respuesta es iniciada por la microglía residente. Sin embargo, al continuar el consumo de grasas en la dieta, las células inmunitarias periféricas, como macrófagos y neutrófilos superan en número gradualmente a la microglía del ARC (Valdearcos et al., 2017), como consecuencia del aumento de la permeabilidad de la BHE en estados de inflamación sistémica crónica (Kigerl, de Rivero Vaccari, Dietrich, Popovich, & Keane, 2014). TLR4, el PRR que se activa en respuesta a LPS también es activado en respuesta a AGS (Huang et al., 2012). En general, en los ratones, TLR4 tiene un nivel de expresión bajo en el cerebro, incluido el hipotálamo, sin embargo, su la expresión aumenta considerablemente cuando los roedores se alimentan con una DAG. Los inductores más potentes de la activación de la vía TLR4/MyD88 hipotalámicos son los AGS de cadena larga, lo que resulta en la activación de la cascada de señalización intracelular que induce la respuesta inflamatoria y determina la resistencia a las señales anorexígenas. Además, la inducción de NF-κB y JNK en la microglía provoca estrés en el retículo endoplásmico (Zhang et al., 2017).
Cuando los AGS en la dieta se sustituyen por cantidades similares de ácidos grasos insaturados (aceite de semilla de lino y aceite de oliva), la inflamación hipotalámica disminuye al igual que la actividad de JNK y NF-kB (Cintra et al., 2012). Por otro lado, la inhibición farmacológica o genética de TLR4 reduce la inflamación del hipotálamo generada por la dieta y protege a los ratones de obesidad e intolerancia a la glucosa (Milanski et al., 2009). Es importante señalar que es la activación del TLR4 derivada del consumo de la dieta y no la obesidad per se la que induce la activación de la microglía del hipotálamo; esto se concluye de los resultados obtenidos con ratones genéticamente obesos ob/ob (deficientes en leptina), db/db (deficientes en el receptor de leptina) y mutantes MC4R-KO (deficientes en el receptor de melanocortina 4), que no presentan activación de la microglía del hipotálamo (Gao et al., 2014). Si bien, la respuesta de la microglía tiene una gran influencia sobre la vigilancia inmunitaria y la respuesta ante alteraciones en la homeostasis del SNC, no es la única célula de la glía que puede influir en los procesos hipotalámicos, ya que se ha demostrado que los astrocitos también son importantes en el control de la conducta dietaria y el metabolismo energético.
Astrocitos y la alteración en la homeostasis energética
Durante los cambios bioquímicos y activación celular que provoca la obesidad en el hipotálamo, además del microambiente proinflamatorio microglial, también se destaca la presencia del fenotipo reactivo de los astrocitos, alteraciones en la citoarquitectura y la interacción sináptica de los circuitos hipotalámicos y la angiogénesis, fenómenos que no se encuentran en otra parte del cerebro, lo que involucra directamente a la influencia del astrocito en estadio reactivo como participante en la desregulación metabólica hipotalámica (González-García & García-Cáceres, 2021). El hipotálamo se posiciona adyacente a la EM, un órgano circumventricular con menores restricciones que la BHE para el ingreso de nutrientes y moléculas extraparenquimales al SNC, lo que obliga a los astrocitos a estar metabólica y responsivamente más activos con respecto a sus homólogos de otras áreas neuroanatómicas (Rodríguez, Blázquez, & Guerra, 2010). Durante el desarrollo de neuroinflamación asociada a DAG, el astrocito cambia su fenotipo de quiescente a “reactivo”, respondiendo y adaptando su morfología y perfil de expresión celular al proceso fisiopatológico dirigido por la inflamación crónica de bajo grado inducida por la obesidad y la neuroinflamación de novo dirigida por la microglía M1, evento generalizado como astrogliosis reactiva (González-García & García-Cáceres, 2021). En la astrogliosis reactiva inducida por DAG, existe una sobrerregulación de la proteína ácida fibrilar glial (GFAP) y un perfil similar a M1, que algunos autores denominan como A1 en homología a la microglía (Liddelow et al., 2017). Los astrocitos A1 producen y liberan citocinas proinflamatorias como TNF-α e IL-6, además adquieren una morfología hipertrófica sin proliferar, pero sobre regulando la vía astrocítica de NF-kB-IkBα (Buckman et al., 2015; Douglass, Dorfman, Fasnacht, Shaffer, & Thaler, 2017). Mecanísticamente, TLR4, al igual que en microglía, induce esta activación proinflamatoria por el reconocimiento de los AGS, potenciado por la recepción de los estímulos inflamatorios producidos por la microglía (González-García & García-Cáceres, 2021), formando un circuito neuroinflamatorio positivo con la microglía y los leucocitos infiltrantes. Además de contribuir al ambiente proinflamatorio, el astrocito reactivo es capaz de desencadenar la expresión de factores solubles que promueven la supervivencia neuronal, en un mecanismo compensatorio predominantemente dirigido por el factor neurotrófico derivado de cerebro (BDNF), favoreciendo la desregulación del control metabólico central a largo plazo, pero evitando la consecuente muerte colateral de las neuronas hipotalámicas (Zhang et al., 2017). Diferentes factores derivados de la lipotoxicidad, obesidad e inflamación crónica de bajo grado afectan tanto a los astrocitos como a la microglía y se recopilan algunos en la tabla 2.
Tabla 2 Moléculas relacionadas con la activación proinflamatoria glial en la obesidad.
| Molécula derivada de la periferia | Origen o causa | Glia que afecta | Consecuencia | Modelo | Referencias |
|---|---|---|---|---|---|
| LPS |
|
|
|
|
(Sousa et al., 2018; Wendeln et al., 2018) (Tarassishin, Suh, & Lee, 2014) |
| TNFɑ, IL-1β, IL-6 |
|
|
|
|
(Maldonado-Ruiz, Montalvo-Martínez, Fuentes-Mera, & Camacho, 2017; Wang et al., 2012) (Hyvärinen et al., 2019; Tarassishin, Suh, & Lee, 2014) |
| AGS | Dieta |
|
|
|
(Vinuesa et al., 2019; Z. Wang et al., 2012) (Gupta, Knight, Gupta, Keller, & Bruce-Keller, 2012) |
| Leptina |
|
|
|
|
(Tang et al., 2007) |
| Gluco- corticoides |
|
|
|
|
(Sorrells, Caso, Munhoz, & Sapolsky, 2009; Sorrells, Munhoz, Manley, Yen, & Sapolsky, 2014; Swierczynska et al., 2015) |
En resumen, la inflamación en el ARC inducida por la dieta está marcada por una acumulación de astrocitos reactivos y microglía activada. La microglía activada y los astrocitos reactivos en el ARC actúan como sensores de los niveles de AGS en la dieta y controlan la intensidad de la inflamación. En este entorno, la microglía y los astrocitos median los efectos inductores de estrés provocados por los AGS de la dieta en las neuronas del ARC y reducen su capacidad de respuesta a la leptina, afectando el comportamiento de la ingesta de alimentos. La inflamación metabólica en el ARC, en respuesta al consumo constante y excesivo de grasas saturadas, ocurre más intensamente que en tejidos periféricos, donde está asociada la obesidad (González-García & García-Cáceres, 2021; Valdearcos et al., 2014) (Fig.1.).
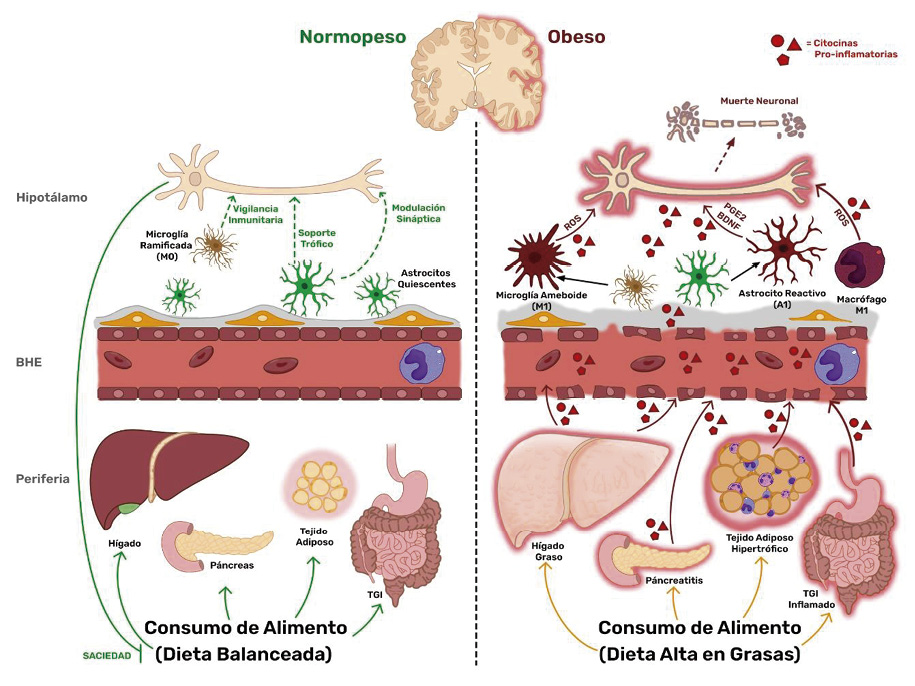
Las neuronas de los núcleos hipotalámicos están inmersas dentro de poblaciones gliales que influencian constantemente su integridad celular. El tipo y cantidad de alimento que se consume tiene influencia directa e indirectamente en el desempeño glial y la función neuronal. La DAG induce la inflamación y deteriora rápidamente la red compleja de los diferentes tipos de células en los órganos periféricos como hígado, tejido adiposo, páncreas y el tracto gastrointestinal (TGI), entre otros, activando sus respectivos sistemas inmunitarios locales y promoviendo inflamación crónica. La DAG deteriora la función e integridad de la BHE a nivel de células endoteliales y glía limitante por el constante estímulo inflamatorio, comprometiendo su función de barrera. La liberación de citocinas proinflamatorias de la microglía M1, astrocitos reactivos, macrófagos perivasculares M1 y de las células inmunitarias infiltrantes, alteran la función de esta red celular, lo que se traduce en la modificación del comportamiento de alimentación y el gasto de energía.
Figura 1 Neuroinflamación hipotalámica asociada a la obesidad
Conclusiones
Con las investigaciones más recientes, se ha podido integrar la información que evidencia al hipotálamo como el principal centro integrador de señales relacionadas con los comportamientos de hambre y saciedad, clave en la regulación del equilibrio energético.
La alteración en el comportamiento alimenticio relacionado a obesidad induce efectos tempranos provocados por la inflamación en el hipotálamo, los cuales preceden a los eventos inflamatorios en tejidos periféricos. La sobre ingesta prolongada conduce a un proceso inflamatorio sostenido del hipotálamo a través de la interacción entre poblaciones de células neuronales y no neuronales como las células gliales, la microglía y los astrocitos. Estas células en el hipotálamo orquestan varias de las funciones metabólicas, este proceso es inicialmente reversible, pero puede desembocar en el desacoplamiento entre la ingesta calórica y el gasto energético, fomentando la sobrealimentación y el aumento de peso.
Además de los ácidos grasos, varios estudios han sugerido que la hiperglucemia y la exposición a la fructosa también pueden inducir inflamación, lo que plantea la posibilidad de que el exceso de nutrientes en sí mismo pueda ser el principal impulsor del proceso inflamatorio. Además, el estudio de los mecanismos de la neuroinflamación asociada a la obesidad puede proporcionar información clínicamente relevante sobre el papel de la inflamación en la patogénesis de los trastornos neurodegenerativos que también se han relacionado con el exceso de grasas en la dieta, como la enfermedad de Alzheimer o la enfermedad de Parkinson.

