










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.56 n.3 Ciudad de México Jul./Sep. 2012
Artículo
The Role of the CH/π Weak Interaction in the Geometrical Conformation: An Aromatic Acetamide Derivative System as an Example
José-Zeferino Ramírez,1 Rubicelia Vargas,1* Itzia I. Padilla-Martínez,2 Anaid G. Flores-Huerta1 and Jorge Garza1
1 Departamento de Química, División de Ciencias Básicas e Ingeniería. Universidad Autónoma Metropolitana-Iztapalapa. San Rafael Atlixco 186, Col. Vicentina. Iztapalapa. C. P. 09340. México D. F. México. ruvf@xanum.uam.mx
2 Departamento de Ciencias Básicas de la Unidad Profesional Interdisciplinaria de Biotecnología del IPN, Av. Acueducto s/n Barrio la Laguna Ticomán, México D.F. 07340, México.
Received January 06, 2012.
Aaccepted March 26, 2012.
Abstract
In this work, three conformers of an aromatic amide derivative are theoretically analyzed. The theoretical methods used were based on the Kohn-Sham version of the density functional theory, considering three exchange-correlation functionals of different types: PBE, TPSS and B3LYP. The results obtained using these methods were compared to those obtained by the many-body perturbation theory to second order (MP2). All these methods where coupled with the 6-311++G(d,p) basis set. The X-ray structure was used as a starting point in the conformational search, as all the methods considered in this work had predicted that this structure would be the conformer with the highest energy, thus obtaining the first important result for this system. The second most important result discovered in this work refers to the large differences found in the predicted structures when applying DFT methods, as compared to the MP2 method. We attribute such differences to dispersion terms not included in the exchange-correlation functionals considered; such a hypothesis is corroborated when a model system (stabilized by dispersion effects) is analyzed by applying the four theoretical methods. By incorporating dispersion effects with the exchange-correlation functional, we found they compared more favorably with the wave-function correlated method.
Key words: Weak Interactions, Dispersion Forces, Grimme Correction, C-H...π Interaction, DFT, MP2, Amide Derivatives.
Resumen
En este trabajo se presentan los resultados del estudio teórico de tres confórmeros de un derivado de una amida aromática. Los métodos teóricos usados están basados en la versión de Kohn-Sham dentro de la teoría de funcionales de la densidad (TFD), se consideran tres funcionales de intercambio y correlación de diferente tipo: PBE, TPSS y B3LYP. Los resultados de estos tres métodos, se comparan con los obtenidos por la teoría de perturbaciones de muchos cuerpos a segundo orden (MP2). Todos los métodos se combinaron con el conjunto de base 6-311++G(d,p). La estructura de rayos X se usó como punto de partida en la búsqueda conformacional, todos los métodos usados predicen ésta como la conformación con la energía más alta, éste es un primer resultado interesante sobre el sistema estudiado. El segundo resultado importante de este trabajo se refiere a las diferencias entre las estructuras predichas por TFD y las obtenidas con MP2. Nosotros atribuimos tales diferencias a los términos de dispersión que no son correctamente considerados en los funcionales de intercambio y correlación; tal hipótesis se corrobora cuando analizamos un sistema modelo (estabilizado por efectos de dispersión) aplicando los cuatro métodos teóricos mencionados arriba. Incorporando los efectos de dispersión en los métodos de TFD, encontramos que comparan favorablemente con el método correlacionado de función de onda.
Palabras clave: Interacciones débiles, fuerzas de dispersión, corrección de Grimme, interacción C-H...π, DFT, MP2, derivados de amidas.
Introduction
Amide molecules are significantly important in biological systems, as they contribute to the backbone of protein molecules [1]. Amides are particularly important in this regard, as the NH group acts as a good hydrogen bond donor with weak acid properties. Secondary amide NHCO groups generally adopt the trans configuration, which facilitates linear-chain networks suitable for forming important supramolecular structures [2-4]. In particular, acetamide clusters have been employed in order to comprehend the cooperativity of hydrogen bonding interactions [5-10], because they serve as simple model systems mimicking the hydrogen bonds, observed in polypeptides and proteins. Recently, acetamide derivatives have also been studied as anthel-mintic agents [11], anti-tuberculosis drugs [12], antioxidants and potential anti-inflammatory substances [13], all of them substituted acetamide with aromatic rings. In all these cases the conformation adopted by each molecule may determine the interaction with the target and their selection of it; consequently the conformation adopted will define their biological activity.
In this sense, a conformational study of acetamide derivatives is interesting in terms of many fields of chemistry; as depending on the geometry, the possibility of forming hydrogen bonds may change, and this would result in different associations and likewise different chemical and physical properties.
Lately, computational chemistry has been widely used to explain the chemical and physical properties of materials, using electronic structure as a starting point. In particular, the application of different approaches within Density Functional Theory (DFT) has proved successful for this purpose [14, 15]. However these approaches manifest certain handicaps known to experts in these theories, who have made several proposals for overcoming these problems; for example it is known, that many of most popular exchange-correlation functionals fail to correctly predict long-range interactions [16]. This problem is not only important in terms of the formation of dimers or adducts, but also concerning the correct description of geometrical molecular conformations. In particular, it has been shown that the weak intramolecular CH/π hydrogen bond [17] plays a significant role in organic chemistry conformations [18-20] and crystal packing [21]. In typical CH/π hydrogen bonds involving sp3- and sp2-CH groups as the hydrogen donor, the stabilization of the complexes (between 1.5 to 2.5 kcal mol-1) is derived from dispersion forces.
In this work, a theoretical study of the molecular structure of the N-(2-benzoylphenyl) acetamide (Figure 1) is carried out. In our study, the intramolecular weak CH/π interaction is the main focus along with its role in affecting the predicted geometrical conformation in gas phase; the experimental X-ray structure has been reported previously and is also discussed here [22]. Naturally, we expect weak energy interactions in the case of this kind of contact and thus we propose the methane-benzene interaction as a model for studying dispersion effects and their consequences on our molecular system.
Methodology
Three exchange-correlation functionals, of varied type have been considered in this work: PBE [23], B3LYP [24] and TPSS [25]. Additionally, the dispersion correction proposed by Grimme [26] has been included in the three exchange-correlation functionals (DFT-D); in fact we had to use a hybrid density functional where the Grimme parameters were defined (B3LYP). The Moller-Plesset many-body perturbation approach to second order (MP2) [27] has been considered, in order to compare the DFT results with other quantum chemistry techniques. All these methods have been coupled with the 6-311++G(d,p) basis set [28]. The reason for this basis set is based on the knowledge of the impact of diffuse functions on the hydrogen bond description and the computational cost implied when applying the MP2 method, so this basis set provides a good balance between these two elements. The methane-benzene adduct was used as a model of the C-H...π interaction presented by the molecule in this study. The curve of the interaction energy between benzene and methane was constructed by single point energies, computed using the same three DFT exchange-correlation functionals mentioned above and the same basis set, the Grimme correction was also included. In the adduct, one of the methane hydrogen is pointing towards the center of the benzene ring, as predicted for other methods described in the literature. At least, eleven single point energies were used for each DFT exchange- correlation functional and DFT-D computations in the interaction curve. All computations were carried out employing the NWChem program [29].
Results
The three most stable structures identified for the N-(2-benzoyl-phenyl)acetamide are presented in Figure 2. In this figure, conformer 1 corresponds to the X-ray experimental structure. From this structure, the other 2 conformers (2 and 3 in Figure 2) were obtained by rotating the dihedral angles displayed in the same figure. The DFT and MP2 methods considered in this work were applied to these structures and a frequency analysis was carried out applying the DFT methods, in order to ensure that these structures are minima on the potential energy surface. These structures indicate evident differences between conformer 3 and the corresponding experimental information. Whereas the X-ray structure exhibits a compact structure, conformer 3 shows an extended conformation, stabilized by an N-H···O hydrogen bond.

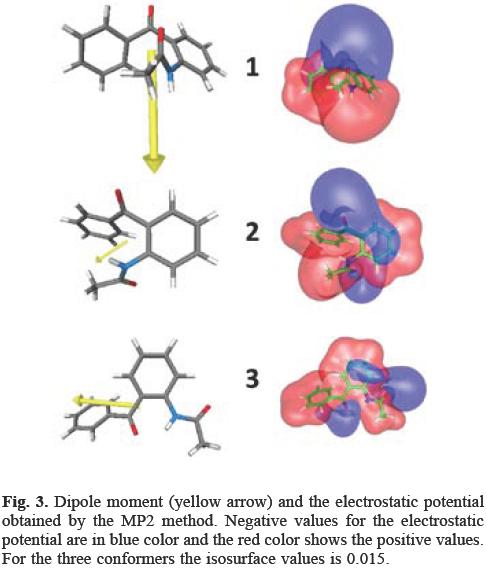
The corresponding relative energies obtained applying the DFT and MP2 methods are presented in Table 1. From this table, we can conclude that the most stable structure in gas phase does not correspond to that found in solid phase; in fact the structure found in the crystal shows the highest energy among the three conformers considered in this work, although MP2 does not connote a substantial energy difference between the three conformers. One reason for the discrepancy between the gas and solid phases is related to the forces necessary for the packing in the solid phase. By evaluating the dipole moment (μ)for the three conformers, it is apparent that conformer 3 connotes the greatest value for this property (in the MP2 method μ is valued at: 7.11 D for 1, 2.20 D for 2 and 4.26 D for 3). The representation of the dipole moment vector for the three conformers is depicted in Figure 3 and in the same figure the electrostatic potential is also plotted. The blue colour represents negative values for the electrostatic potential whereas the red represents positive values. By analyzing the electrostatic potential, it was found that for conformer 3, the negative values for this quantity (provided mainly by the oxygen atoms) are always opposite to the positive values which induce a large value on the dipole moment. In the case of conformers 2 and 3, the oxygen atoms are opposite with respect to each other and for this reason their dipole moments are not so substantial. The large dipole moment of 1 may be responsible for stacking in the solid phase. Evidently, this proposition must be confirmed by comparing the energies of these conformers using periodic boundary techniques, but this is outside the scope of this study.
The structure of conformer 3 predicted by DFT methods is quite similar to that obtained with MP2. However, the DFT-MP2 difference is more pronounced for conformers 1 and 2, especially for conformer 2 where the RMS is around 0.50 Å when DFT and MP2 structures are compared; in Figure 4, the comparison between the B3LYP and MP2 geometries is presented as an example. It can be seen in this figure that the main difference can be observed in the case of the CH3 group, which is pointing to the benzene ring in the structure predicted by the MP2 method, whereas in the case of the geometry obtained by B3LYP this is not evident. One possible reason of this discrepancy is that the MP2 method is revealing certain weak intramolecular interactions that the DFT approaches are not able to portray. In order to prove this hypothesis, the complex benzene-CH4 was studied here, using the same methods and basis set functions. This complex is very well studied and is known for manifesting an attractive interaction; the preferred geometry configuration depicts one of the hydrogen atoms of the methane pointing to the center of the benzene ring [30]. The estimated energy interaction (ΔEint) for this configuration using CCSD(T) as the basis limit is 1.454 kcal/mol [31].

We tested the methods considered in this work for the benzene-CH4 complex in its preferred configuration; in the diagram of Figure 5, we present only the MP2 and PBE results for the sake of simplicity. As is apparent in this figure, MP2 clearly presents a minimum in the potential energy surface of the benzene-CH4 complex, whereas PBE presents only a slight minimum, in a way similar to that portrayed by TPSS. B3LYP however does not predict an attractive interaction for this complex. This behaviour is due to the lack of dispersion terms in these DFT approaches. However Grimme [26] proposed an empirical correction for this erroneous behaviour, incorporating the dispersion effects on the total energy. In Figure 5, the potential energy surface is presented for the benzene-CH4 complex computed with PBE and corrected by applying Grimme's approach (PBE-D). It is obvious in this figure that this correction improves the benzene-CH4 interaction. We applied this correction as implemented in NWChem code [29] to all DFT methods studied here, in order to estimate the ΔEint for the benzene-CH4 complex, the results are: -1.7 kcal/mol for B3LYP, -1.9 kcal/mol for PBE and -2.0 kcal/mol for TPSS. All of these DFT approaches including the Grimme correction produce an attractive interaction in the benzene-CH4 complex, however even with the dispersion correction, the energy interaction of DFT falls far short of the MP2 prediction (-2.4 kcal/mol). We know that corrections for basis set superposition errors must be implemented in order to adjust our estimations especially in the case of the MP2 method, but this is not the intention of this discussion. We want to emphasize that without the dispersion correction, the DFT approaches do not predict an attractive interaction between a CH3 group and a benzene ring as that presented by the target molecule of our study and this is the reason why MP2 and DFT approaches give different conformations, when these interactions are present. With this result in mind, we re-optimized the 7V-(2-benzoylphenyl)acetamide with all DFT approaches adding the dispersion correction (results in parenthesis in Table 1) and the geometries obtained were in greater conformity with those obtained with MP2.

Analyzing the results in Table 1, the DFT results are found to be consistent, whether or not the dispersion correction is applied (number in parenthesis); all of the DFT relative energies are quite different from those obtained when applying the MP2 method. The role of the dispersion correction on the conformational energies is evident; this correction reduces the relative energies but this reduction is not enough to reach the MP2 values, this may be because MP2 overestimates the stabilization energy of these contacts, as it does in the case of the benzene-CH4 complex.
Conclusions
The gas phase conformation of the 7V-(2-benzoylphenyl)acetamide predicted by theoretical methods is stabilized by a N-H···O hydrogen bond, whereas in the X-ray structure adopted by the molecule it connotes a conformation that may be stabilized by intermolecular electrostatic interactions. The conformer stabilized by this N-H···O hydrogen bond is quite similar in all the theoretical methods used in this work, however the weaker interactions that stabilize other conformers are not predicted in a similar way by MP2 and the DFT approaches, consequently the predicted geometries and conformer stabilization energies do not concur. When a dispersion correction is included in DFT approaches, the geometries become increasingly similar but the relative energies predicted by MP2 and DFT continue to disagree which may be due at least in part, to the fact that MP2 tends to over estimate these weak interactions.
Acknowledgements
This work was carried out at the Laboratorio de Supercómputo y Visualización en Paralelo of the Universidad Autónoma Metropolitana-Iztapalapa. The authors are grateful for financial support of CONACYT, through projects 155070 and 154784.
References
1. Nelson, D. L.; Cox, M. M.; Lehninger, A. Principles of Biochemistry, Worth Publishers, NY, 2000, Ch. 1. [ Links ]
2. Subramanian, S.; Zaworotko, M. J. Coord. Chem. Rev. 1994, 137, 357-401. [ Links ]
3. Aakeröy, C. B.; Seddon, K. R. Chem. Soc. Rev. 1993, 397-407. [ Links ]
4. McDonald, J. C.; Whitesides, G. M. Chem. Rev. 1994, 94, 2383-2420. [ Links ]
5. Mahadevi, A. S.; Neela, Y. I.; Sastry G. N. Phys. Chem. Chem. Phys. 2011, 13, 15211-15220. [ Links ]
6. Papamokos G. V.; Demetropoulos, I. N. J. Phys. Chem. A 2004, 108, 7291. [ Links ]
7. Ludwig, R.; Weinhold, F.; Farrar, T. C. J. Phys. Chem. A. 1997, 101, 8861. [ Links ]
8. Ludwig, R.; Weinhold, F.; Farrar, T. C. J. Chem. Phys. 1997, 10, 499. [ Links ]
9. Hirst, J. D.; Hirst; D. M.; Brooks, C. L. J. Phys. Chem. A 1997, 101, 4821. [ Links ]
10. Wong, M. W.; Wiberg, K. B. J. Phys. Chem. 1992, 96, 668. [ Links ]
11. Sawant, R.; Kawade, D. Acta Pharm. 2011, 61, 353-361. [ Links ]
12. Bai, Y.; Wang, L.; Chen, Y.; Yuan, L.; Xu, W.; Sun T. J. Mol. Struct. 2011, 1005, 113-120. [ Links ]
13. Autore, G.; Caruso, A.; Marzocco, S.; Nicolaus, B.; Palladino, C.; Pinto, A.; Popolo, A.; Sinicropi, M. S.; Tommonaro, G.; Saturnino, C. Molecules 2010, 15, 2028-2038. [ Links ]
14. Parr, R. G.; Weitao, Y. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989. [ Links ]
15. Chatarraj, P. K. Editor. Chemical Reactivity Theory: A Density Functional View; CRC Press: Florida, 2009. [ Links ]
16. Grimme, S. Comput. Mol. Sci. 2011, 1, 211-228. [ Links ]
17. Nishio, M.; Hirota, M.; Umezawa, Y. The CH/π Interaction. Evidence, Nature, and Consequences; Wiley-VCH: New York, 1998. [ Links ]
18. Nishio, M.; Hirota, M. Tetrahedron 1989, 45, 7201-7245. [ Links ]
19. Takahashi, O.; Yamasaki, K.; Kohno, Y.; Kurihara, Y.; Ueda, K.; Umezawa, Y.; Suezawa, H.; Nishio, M. Tetrahedron 2008, 64, 2433-2440. [ Links ]
20. Nishio, M.; Umezawa, Y.; Honda, K.; Tsuboyama, S.; Suezawa, H. Cryst. Eng. Comm. 2009, 11, 1757-1788. [ Links ]
21. Nishio, M. Cryst. Eng. Comm. 2004, 6, 130-158. [ Links ]
22. Gómez, M., Gómez-Castro, C. Z., Padilla-Martínez, I. I., Martínez-Martínez, F. J., González, F. J. J. Electroanal. Chem. 2004, 567, 269-276. [ Links ]
23. (a) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865-3868. [ Links ] (b) Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1997, 78, 1396-1396. [ Links ]
24. (a) Becke, A. D. J. Chem. Phys. 1993, 98, 5648-5652. [ Links ] (b) Becke, A. D. J. Chem. Phys. 1993, 98, 1372-1377. [ Links ]
25. Tao, J.M.; Perdew, J.P.; Staroverov,V.N.; Scuseria, G. E. Phys. Rev. Lett. 2003, 91, 146401(1)-146401(4). [ Links ]
26. Grimme, S. J. Comput. Chem. 2006, 27, 1787-1799. [ Links ]
27. (a) Maller, C.; Plesset, M. S. Phys. Rev. 1934, 46, 618-622. [ Links ] (b) Szabo, A.; Ostlund, N. S. Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory; McGraw-Hill, New York, 1989. [ Links ]
28. Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. J. Chem. Phys. 1980, 72, 650-654. [ Links ]
29. Valiev, M.; Bylaska, E. J.; Govind, N.; Kowalski, K.; Straatsma, T. P.; van Dam, H. J. J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T. L.; de Jong, W. A. Comput. Phys. Commun. 2010, 181, 1477. [ Links ]
30. Tsuzuki, S.; Honda, K.; Uchimaru, T.; Mikami, M.; Tanabe, K. J. Am. Chem. Soc. 2000, 122, 3746-3753. [ Links ]
31. Ringer, A. L.; Figgs, M. S.; Sinnokrot, M. O.; Sherrill, C. D. J. Phys. Chem. A 2006, 110, 10822-10828. [ Links ]
* The asterisk indicates the name of the author to whom inquires about the paper should be addressed.

