











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink

PRESENTACIÓN DEL CASO
Paciente del sexo masculino de 3 años de edad que acudió a centro oftalmológico. Se refirió que desde hacía unos 3 meses habían notado un “reflejo blanco” en el ojo izquierdo en las fotos que le habían tomado. En el interrogatorio dirigido, uno de sus padres mencionó que desde hacía 2 meses había notado que su hijo se acercaba mucho a los objetos o a la televisión.
A la exploración física, se reportó una capacidad visual de 20/400 y 20/20. Segmento anterior de ambos ojos se reportó sin alteraciones. A la búsqueda del reflejo rojo, se encontró ausente el ojo izquierdo. Se realizó exploración bajo dilatación farmacológica. Ojo izquierdo: se observó una lesión blanquecina de bordes mal definidos que abarcó desde meridiano 3 a meridiano 8. Se decidió solicitar una ecografía ocular modo A y modo B. Se reportó lesión ocupante en sector inferior, heterogénea, con eco hiperecogénico intralesional con atenuación posterior, en relación a área de calificación. El eco modo A reportó hiperreflectividad alta. Se decidió realizar estudios de extensión, por lo que se realizó una tomografía computarizada, donde se encontraron datos compatibles con retinoblastoma del ojo izquierdo. Por lo anterior, se decidió realizar enucleación del ojo izquierdo, donde el estudio histopatológico de este paciente reportó retinoblastoma endofítico poco diferenciado (20%).
DISCUSIÓN Y ANÁLISIS DEL CASO
El retinoblastoma es una neoplasia maligna primaria de la retina, que proviene de células inmaduras retinianas1. Es el tumor maligno intraocular más frecuente en la infancia, aunque es un cáncer poco frecuente2, ya que representa el 2-4% de todos los cánceres pediátricos, de ellos 2/3 de los casos son diagnosticados antes de los 2 años de edad; y más de 90%, antes de los 5 años3. La edad media de diagnóstico es a los 12 meses para la presentación unilateral, y 24 meses para la bilateral. Afecta al sexo femenino y masculino con una frecuencia equitativa, sin predilección de raza y con una incidencia mundial de 1:15,000-1:20,000 nacidos vivos2.
Se han identificado dos formas de presentación: unilateral (tumor aislado en un ojo) y bilateral (tumor multifocal en ambos ojos)2.
Se inicia por la mutación en el gen supresor tumoral RB1, lo que lleva a la transformación maligna de las células retinianas primitivas4 y hay dos formas genéticas:
MANIFESTACIONES CLÍNICAS
La leucocoria (brillo blanco en la pupila) es el signo más frecuente y se observa cuando el tumor aún se encuentra contenido dentro del ojo, el retinoblastoma es curable en los primeros 3-6 meses posteriores al primer signo de leucocoria.
La deprivación visual, a causa del tumor intraocular, resulta en estrabismo, ya sea hacia afuera (exotropia) o adentro (endotropia), resultando la segunda manifestación clínica más frecuente. Este signo es ignorado por muchos médicos de primer contacto, refiriéndolo como un problema de desarrollo, que desaparecerá al crecer el niño4.
Otras manifestaciones son ojo rojo, hifema (hemorragia en cámara anterior), neovascularización de iris, que nos indican de inflamación intraocular y disminución de agudeza visual y proptosis5.
DIAGNÓSTICO
El retinoblastoma se debe diagnosticar durante una consulta con el pediatra o el médico general, cuando se busca el reflejo rojo en el fondo de ojo del paciente. Al encontrarse alterado el reflejo, debe ser referido de manera inmediata al oftalmólogo, quien deberá realizar una exploración con oftalmoscopía del fondo de ojo.
DIAGNÓSTICO POR IMAGEN
Un abordaje inicial para este tipo de lesiones puede hacerse buscando las principales etiologías de calcificaciones intraoculares (tabla 1)6, tomando en cuenta para llegar a un diagnóstico de precisión la edad, los antecedentes, las regiones afectadas y la bilateralidad.
Las características por tomografía computada del retinoblastoma son que es una tumoración intraocular unilateral que tiene una mayor densidad en comparación del humor vítreo, y que tras la aplicación de medio de contraste se observa un reforzamiento moderado de la lesión7. Como ya se mencionó, las calcificaciones son un dato importante, ya que sólo cerca del 5-10% de ellos, no muestran esta característica8.
Tabla 1 Causas de calcificaciones intraoculares encontradas por tomografía computada
| Común | Menos común | Raro |
|---|---|---|
| Calcificaciones de Drusen | Retinoblastoma | Osteoma coroideo |
| Ptisis bulbi | ||
| Calcificaciones oculares, etc. |
En la figura 1 y figura 2 se pueden observar las imágenes tomográficas características descritas con el caso clínico presentado.
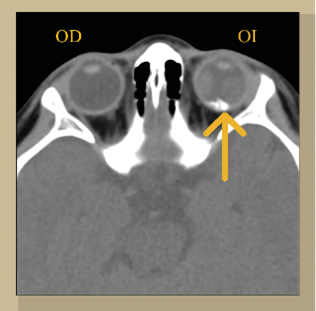
Figura 1 Tomografía transversal de órbitas en fase simple donde se observa en el interior del globo ocular izquierdo (OI), una imagen de morfología irregular, de bordes regulares, bien definidos, isodensas a calcio (flecha), el ojo derecho (OD) se encuentra sin alteraciones.
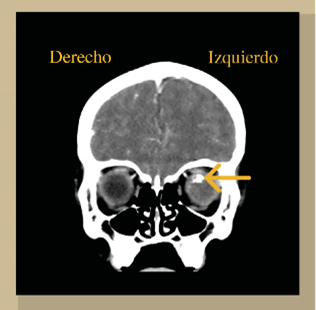
Figura 2 Reconstrucción coronal donde se observa que, tras la administración del medio de contraste, la lesión previamente descrita presenta un reforzamiento nodular heterogéneo. Obsérvese de manera comparativa que el ojo derecho (OD) se encuentra en situación, morfología y tamaño habitual.
Para descartar los demás diagnósticos como las calcificaciones de Drusen, es necesario identificar que en estas se distribuyen a lo largo de la retina y que no hay compromiso del resto de los tejidos blandos, sobre todo en pacientes de mayor edad y con degeneración papilar. En pacientes con antecedentes de traumatismo, infección, inflamación o radiación se podría llegar a pensar en la ptisis bulbi, pero contrario al retinoblastoma, el globo ocular y el nervio óptico se encuentran disminuidos de tamaño o atróficos. A pesar de ser un diagnóstico raro, es necesario descartar la posibilidad de un osteoma coroideo en mujeres jóvenes y con pérdida progresiva de la visión, donde la calcificación tiene morfología laminar y se localiza en la región posterior y yuxtapapilar, con reforzamiento moderado tras medio de contraste.
A pesar de que la tomografía computada se ha considerado el método de elección para estudiar las calcificaciones, actualmente se prefieren otros para evitar los riesgos de la exposición a la radiación. Por lo que la combinación del ultrasonido y la resonancia magnética (RM) ya se consideran la primera línea de diagnóstico por imagen9.
La RM es de mucha utilidad para evaluar la extensión local, donde en T1 presenta una señal isointensa o levemente hiperintensa en relación con el humor vítreo y en T2 se comporta como una lesión levemente hipointensa, las cuales son de mucha utilidad para evaluar la extensión al nervio óptico, cámara anterior y grasa periorbitaria7.
Recientemente existen otras secuencias de RM que nos ayudan a estudiar con gran precisión las calcificaciones de las lesiones, pero sin el riesgo que la radiación conlleva, como T2*WI (T2* weighted imaging) y como susceptibilidad magnética (susceptibility-weighted imaging)9.
Además, debemos de evaluar de manera sistemática otras estructuras en busca de una posible existencia de una lesión contralateral, compromiso de la glándula pineal o tumoración supraselar6.
A pesar de estas recomendaciones, cada centro hospitalario debe tomar la mejor decisión, de acuerdo a los métodos diagnósticos existentes en su hospital, costos e infraestructura del mismo, para poder ofrecer la opción ideal para cada paciente.
RADIOGENÓMICA
En años recientes, ha surgido una nueva línea de investigación que combina los fenotipos por imagen y la genómica, a esto se le ha denominado imagen genómica o radiogenómica10. Específicamente para el retinoblastoma, se han identificado ciertos perfiles de expresión genética, los cuales pueden ayudar a predecir las características moleculares como la pérdida de los fotoreceptores, localización, márgenes, patrón de crecimiento, etc. que podría implicar un cambio directo en la toma de decisiones y en tratamientos futuros de esta enfermedad11.
TRATAMIENTO
El principal objetivo es salvar la vida del niño, seguido por la preservación del ojo y la visión. Existen muchas opciones, dependiendo de la lateralidad y extensión.
La enucleación es el tratamiento definitivo para un retinoblastoma unilateral avanzado, siembras vítreas o desprendimientos de retina4.
La quimioterapia se indica en pacientes con enfermedad extraocular, bilateral o en pacientes con enfermedad unilateral en los que se desea preservar el ojo3.
La radioterapia se aplica en los pacientes que no han tenido respuesta a la quimioterapia o terapias locales4.
Las terapias locales (crioterapia, láser) se aplican en tumores < 3-6mm, en pacientes con tumores bilaterales3.
PRONÓSTICO
El tratamiento y pronóstico depende del estadio al inicio de la presentación. Algunos predictores son el tamaño del tumor, localización, presencia de líquido subretiniano, siembras y las características histopatológicas4.
SEGUIMIENTO
Los pacientes se mantienen en vigilancia a las 2-4 semanas posteriores al tratamiento. Al encontrarse certeza de la erradicación del tumor, posteriormente cada 3 meses por 2 años y a continuación cada 6 meses hasta cumplir 6 años, hasta finalmente anual1.

