











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El síndrome hemofagocítico es una enfermedad amenazante para la vida que debe ser sospechada en aquellos pacientes con fiebre y citopenias. Tradicionalmente se clasifica como primario, observado principalmente en niños y debido a alteraciones genéticas, o secundario a procesos infecciosos o neoplásicos; en el caso de estar asociado a enfermedades reumatológicas se conoce como síndrome de activación de macrófagos1. Existen pocos estudios epidemiológicos acerca de esta enfermedad, sin embargo, se estima que la incidencia a nivel mundial es de 1.2 casos por cada millón de habitantes, aunque esta cifra puede estar subestimada debido a la dificultad para realizar el diagnóstico y a que los síntomas y signos de la enfermedad son poco específicos2.
Desde 1991 la sociedad del histiocito publicó una serie de 6 criterios diagnósticos que se debían cumplir para confirmar el cuadro, que fueron revisados en el 2004, y se agregaron 3 criterios adicionales con el objetivo de estandarizar el diagnóstico y no subestimar la enfermedad (tabla 1). Un diagnóstico oportuno permite brindar una terapéutica eficaz lo cual disminuye significativamente la mortalidad de la enfermedad3.
CASO CLÍNICO
Paciente del sexo masculino, de 40 años, sin antecedentes de importancia. Inició su padecimiento una semana previa a su ingreso con un cuadro caracterizado por cefalea frontal, opresiva, de inició súbito, sin irradiaciones, sin atenuantes ni exacerbantes, sin dolor retroorbitario; lo describió como el peor de su vida, sin náusea, fotofobia, fonofobia, datos de focalización; posteriormente se agregaron picos febriles no cuantificados asociados a datos de bacteremia (diaforesis profusa de predominio nocturno), por lo que recibió tratamiento con cefixima por 4 días, paracetamol e ibuprofeno, sin mejoría de la sintomatología, razón por la que decidió acudir a valoración a Urgencias, en donde se le administraron analgésicos para el control del dolor, se realizó una tomografía axial computada (TAC) de cráneo, la cual no muestra alteraciones; de sus estudios de laboratorio destaca una leucopenia y un patrón colestásico en las pruebas de función hepática (PFH) (tabla 1). Se realizó una punción lumbar, que resultó sin alteraciones en el citoquímico y citológico, y sin crecimiento en el cultivo. Se inició con ceftriaxona por parte de infectología, por sospecha de fiebre tifoidea.
Tabla 1 Estudios de laboratorio
| 23/01/2016 | Hb: 13.4, Hto 40.5, VGM: 94.2, HCM: 31.3; CMH: 33.2, ADE: 15, Plaq.: 328, Leu.: 15.7, Neu. ABS: 11.9, Linf. ABS: 2.1, MONO ABS: 1.4, EOS ABS: 0.1, BUN: 15.3, urea: 32.7, CRES: 0.83, BT: 0.99, BD: 0.27, BI: 0.72, ALT: 136; AST: 78, FA: 141, GGT: 221 DHL: 183, ferritina: 893.8, influenza A y B: negativos LCR: incoloro, transparente, células 0, eritrocitos 0, crenocitos 0, GLU 56.8, prot. 28.4, Gram: negativa, tinción china: negativo |
| 24/01/2016 | Hb: 12, Hto 34.9, VGM: 93.4, HCM: 32.1, CMH: 34.3 ADE: 13.7, Plaq.: 105 Leu.: 3, Neu. ABS: 2.3, Linf. ABS: 0.3, TP: 10.9, INR: 0.99, PT: 5.86, ALB: 3.8, GLO: 2.1, BT: 2.82, BD: 0.72, BI: 2.1, ALT: 88, AST: 103, FA: 162 GGT: 218 DHL: 549 hemocultivos negativos |
| 25/01/2016 | Hb: 12.1, Hto: 35.6, VGM: 95.2, HCM: 32.4, CMH: 34, ADE: 14, Plaq.: 90, Leu.: 3.4, Neu. ABS: 2.7, Linf. ABS: 0.3, TP: 11.4, INR: 1.03, VSG: 30, PT: 4.85, ALB: 2.71, GLO: 2.1 BT: 4.28, BD: 2.49, BI: 1.79, ALT: 125, AST: 143, FALC: 210, GGT: 187, DHL: 523 PCR: 154.63. Reacciones febriles: parafítico A y B, tifico O H brucella avortus: negativo, proteus OX: positivo 1:40 |
| 26/01/2016 | Hb: 11.3, Hto: 33, VGM: 94.1, HCM: 32.2, CMH: 34.2, ADE: 14.3, Plaq.: 82, Leu.: 2.9 Neu. ABS: 2.1, Linf. ABS: 0.5, MONO ABS: 0.3, PT: 4.45, ALB: 2.42, GLO: 2, BT: 4.92, BD: 3.2, BI: 1.7, ALT: 114, AST: 124, FA: 252, GGT: 167, DHL: 465. Perfil de hepatitis A, B Y C: negativo |
| 27/01/2016 | Hb: 11.4, VGM: 94.2, HCM: 32.3, CMH: 34.3, Plaq.: 97, Leu.: 3, Linf. ABS: 0.7, PT: 4.6, ALB: 2.31, GLO: 2.3 BT: 4.97, BD: 3.11, BI: 1.86, ALT: 100, AST: 118, FA: 303, GGT: 168, DHL: 40. AMO: leucemia/linfoma inmunofenotipo (llib. Antígenos CD2, CD3, CD5, CD7, CD10, CD11B, CD13, CD14, CD15, CD19, CD20, CD33, CD34, CD38, CD45, CD64, CD117, CD79A, HLA-DR, MPO Y TDT. Patrón de maduración de la serie granulocitica, similares a un síndrome mielodisplasico. Mielocultivo: sin desarrollo de microorganismos a los 28 días de incubación y Baar negativo a 8 semanas de incubación |
| 29/01/2016 | Hb: 10.9, VGM: 92.9, HCM: 31.9, CMH: 34.3, ADE: 14.1, Plaq.: 113, Leu.: 4.8, Neu. ABS: 2.6, Linf. ABS: 1.1, MONO ABS: 0.8, TP: 10.5, INR: 0.96, TT: 16.6, fibrinógeno: 333, reticulocítos: 3.4%, retis ABS: 117, BUN 15.1, urea: 32.3, CRS: 1.31, DD: 1740 triglicéridos: 370.5, PT: 4.69, ALB: 2.32, glob: 2.4, BT: 3.36, BD: 1.91, BI: 1.45, ALT: 84, AST: 93, FA: 594, GGT: 240, DHL: 319, ferritina: 4195, VIH: no reactiva |
| 30/01/2016 | Hb: 10.8, Hto: 32, VGM: 95.1, HCM: 32.1, CMH: 33.8, ADE: 15.1, Plaq.: 150 Leu.: 7.3 Neu. ABS: 4.2, Linf. ABS: 2.1, TP: 10.3, INR: 0.94, PT: 5.12, ALB: 2.49, GLO: 2.6, BT: 2.09, BD: 0.95, BI: 1.14, ALT: 87, AST: 65, FA: 587, GGT: 230, DHL: 281. Anticuerpos antinucleares: positivos patrones moteados (1:160), DNA doble: negativo |
| 31/01/2016 | Hb: 10.3, Hto: 30.9, VGM: 96.3, HCM: 32.1, CMH: 33.3, ADE: 15.1, Plaq.: 178, Leu.: 6.9, Neu. ABS: 4.3, Linf. ABS: 2, TP: 10, INR: 0.91, PT: 5.27, ALB: 2.56, GLO: 2.7, BT: 1.76, BD: 0.75, ALT: 80, AST: 52, FA: 509, GGT: 205, DHL: 255. |
| 02/02/2016 | Hb: 10.5, VGM: 95.7, Leu.: 7.2, Neu. ABS: 4.6, Linf. ABS: 1.9, TP: 11.3, INR: 1.03, BUN: 18.2, urea: 38.9, CRS: 0.83, ALB: 2.66, GLOB: 2.4, BT: 1.56, BD: 0.54, BI: 1.02, ALT: 71, AST: 38, FA: 401, GGT: 201, DHL: 201. Anticuerpos de Epstein Barr: cápside IGG: 485, cápside IGM: <10, AC IGG temprano: 5, antígeno IGG nuclear: 344, DNA Epstein Bar >200, parvovirus B19 < 100. |
| 05/02/2016 | PCR de virus respiratorios: influenza A positivo. Parainfluenza tipo 4, adenovirus, parainfluenza tipo 1, tipo 2, tipo 3, influenza B, rinovirus A/B/C, enterovirus metapneumovirus, bocavirus 1/2/3/4, coronavirus, virus sincitial respiratorio A y B negativos. Lavado bronquio alveolar de lóbulo superior izquierdo: BAAR negativo. Cultivo respiratorio: sin aislamiento, hongos y micobacterias. Cultivo micobcaterias negativo. PCR en tiempo real para mycobacterium no tuberculosis negativo |
| 16/02/2016 | Hb: 13.4, Hto: 40.5, VGM: 94.25, HCM: 31.3, ADE: 15, Plaq.: 328, Leu.: 15.7, Neu. ABS: 11.9, Linf. ABS: 2.1, MONO ABS: 1.4, EOS ABS 0.1, TP: 11, INR:1, BUN: 15.3, CRS: 0.83, PT: 3.52 ALB: 2.99, GLOB: 0.5, BT: 0.99, ALT: 136, AST: 78, GGT: 221, DHL: 183, ferritina: 893.8. |
A la exploración física inicial destacó la presencia de adenopatías múltiples cervicales, pequeñas, móviles, no dolorosas a la palpación, así como hepatomegalia de 2 cm por debajo del reborde costal, no dolorosa. Se descartó hepatitis, mononucleosis infecciosa, VIH y no se documentó crecimiento bacteriano en los cultivos. Debido a la persistencia de fiebre y citopenias se solicitó analizar ferritina y triglicéridos, los cuales se encontraron elevados, por lo que cumplía criterios para síndrome hemofagocítico.
Fue valorado por hematología, quienes realizaron aspirado de médula ósea, donde se observó una médula hipercelular (celularidad del 80%) con necrosis focal, detención de la diferenciación eosinofílica en la serie mieloide, no se observaron microorganismos patógenos ni neoplasias (figuras 1a y 1b), se complementó con una tomografía por emisión de positrones-tomografía computada (PET/TC) con resultado de metabolismo glucolítico anormal en ganglios linfáticos mediastinales, hepatoesplenomegalia con inversión de la actividad hígado/bazo.

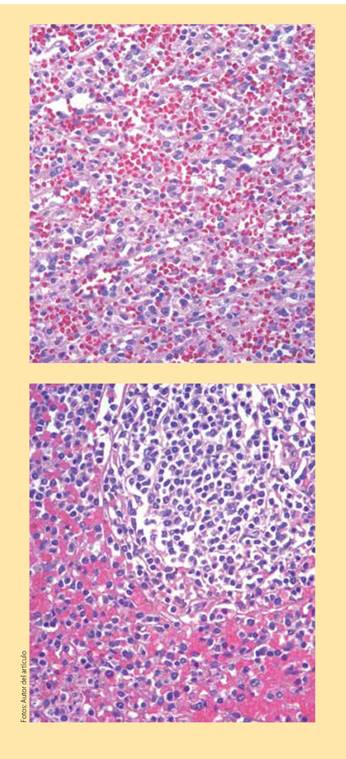
Fotos: Autor del artículo
Figura 1ay 1b Aspirado de médula ósea: medula ósea hipercelular (celularidad del 80%) con necrosis focal, detención de la diferenciación eosinofílica en la serie mieloide, no se observan microorganismos
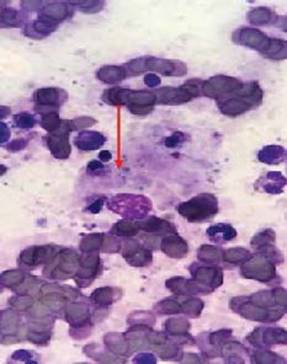
Se realizó esplenectomía cuyo reporte histopatológico muestra pulpa roja con congestión pasiva crónica, plasmocitosis reactiva y megacariocitos normales multifocales, sin presencia de microorganismos patógenos, granulomas ni neoplasia (figuras 2a y 2b). Como parte del protocolo diagnóstico de síndrome hemofagocítico se realizó una biopsia de ganglio linfático mediastínico, en donde se reportaron macrófagos con células sanguíneas intracitoplasmáticas, compatible con hemofagocitosis (figura 3). Debido a estos hallazgos inició tratamiento con dexametasona 16 mg IV al día y etopósido, recibiendo 8 semanas de tratamiento. El paciente presentó mejoría clínica y de sus estudios de laboratorio, sin documentarse recaída de la enfermedad.
Fotos: Autor del artículo
Figura 2a y 2b Pulpa roja con congestión pasiva crónica, plasmocitosis reactiva y megacariocitos normales multifocales. No se identificaron microorganismos patógenos, granulomas ni neoplasia
DISCUSIÓN
El síndrome hemofagocítico es una enfermedad que se produce por una activación excesiva del sistema inmunológico la cual resulta ineficaz. Puede ser una enfermedad grave, amenazante para la vida y de difícil diagnóstico para el médico clínico. Es una enfermedad con una incidencia baja, aproximadamente se reportan 1.2 casos por cada millón de personas por año, aunque se cree que se encuentra infradiagnosticada. Típicamente se divide en 2 grupos: primaria y secundaria4.
En cuanto a la forma primaria, conocida como linfohistiocitosis hemofagocítica familiar autosómica recesiva (FHL), se desconoce su incidencia, ya que todos los estudios epidemiológicos abarcan a los casos secundarios. De acuerdo con la alteración genética, se subdivide en 5 síndromes: FHL 1, FHL 2, FHL 3, FHL 4 y FHL 5; habitualmente se detecta en niños, aunque hay casos aislados reportados en los que se documenta por primera vez en la adultez. Las mutaciones en los genes producen defectos en la regulación inmune, principalmente en los linfocitos T citotóxicos y linfocitos NK. La mutación más común es la encontrada en el gen de la perforina, siendo ésta un importante factor en la modulación inmune y en la apoptosis5. En la tabla 2 se describen las alteraciones genéticas de los 5 subtipos.
Tabla 2 Alteraciones genéticas de los 5 subtipos FHL
| Subtipo de FHL | Gen |
|---|---|
| 1 | Desconocido |
| 2 | PFR1 |
| 3 | UNCI3D |
| 4 | STX11 |
| 5 | STXBP2 |
En cuanto a la forma secundaria, existe una serie de condiciones que la desencadenan, siendo las infecciones las responsables del 50% de los casos (la mayoría asociada a infecciones virales), seguido de malignidad, enfermedades reumatológicas y síndrome de inmunodeficiencia6. En cuanto a las causas infecciosas es importante descartar infección por el grupo de herpes virus, parvovirus B19, VIH y hepatitis B. Las causas infecciosas no virales que más se asocian a este síndrome son leishmaniasis y malaria6. Entre las causas malignas, las neoplasias hematológicas son las más comunes, y entre las enfermedades reumatológicas, el lupus eritematoso sistémico es el de mayor prevalencia7.
Se desconoce la patogenia exacta de la enfermedad, sin embargo, se han identificado ciertas características que podrían explicar sus síntomas: existe una hiperactivación de macrófagos con una hipersecreción de citocinas, lo cual puede explicar el daño a ciertos órganos; también se cree que existe una eliminación ineficaz de los antígenos, lo que conlleva a una estimulación inmune y una hemofagocitosis inapropiada. Existe también una activación excesiva de los linfocitos T CD8, que se puede medir a través del CD25 con una actividad disminuida de los linfocitos NK5.
Esta enfermedad se caracteriza principalmente por fiebre, esplenomegalia y citopenias, las cuales pueden asociarse a hipertrigliceridemia, hipofibrinogenemia con coagulopatía, afección hepática caracterizada por elevación de las transaminasas y síntomas neurológicos como convulsiones, ataxia y alteraciones del estado mental. Una de las cohortes más grandes, encontró que existen ciertas características clínicas y laboratoriales que están presentes en los pacientes y no se encuentran incluidas dentro de los criterios diagnósticos como, por ejemplo: hepatomegalia (encontrada en el 95% de los casos), adenopatías (encontrada en el 33% de los casos), elevación de transaminasas (encontrada en el 76% de los casos) y alteraciones neurológicas (encontradas en el 33% de los casos). Además, se puede encontrar rash, edema, ictericia, hipoalbuminemia, hiponatremia e hiperproteinorraquia4.
El diagnóstico se estandarizó en el 2004 y puede realizarse a través de 2 formas:
Tabla 3 Criterios diagnósticos de 2004 del síndrome hemofagocítico
| Característica | Corte |
|---|---|
| 1. Fiebre | |
| 2. Esplenomegalia | |
| 3. Citopenia
|
Afección mínima
de 2 líneas celulares Menor a 9 g/L Menor a 100 ( 109/L Menor a 1 ( 109/L |
| 4. Hiperferritinemia | Mayor a 500 µg/L |
| 5. Hipofibrinogenemia O hipertrigliceridemia | Menor a 1.5 g/L
Mayor a 265 mg/dL |
| 6. CD25 soluble | Mayor a 2400 UI/mL |
| 7. Actividad celular de las NK disminuida | Referencia de acuerdo con cada laboratorio |
| 8. Hemofagocitosis en médula ósea, ganglios linfáticos o bazo | |
A pesar de la estandarización de los criterios diagnósticos, el diagnóstico de síndrome hemofagocítico suele realizarse de forma tardía, por lo que una vez establecido es importante identificar si el paciente se encuentra clínicamente estable o críticamente enfermo, para brindar una terapéutica adecuada.
En caso de que se sospeche que el síndrome hemofagocítico es desencadenado por una infección, se debe brindar tratamiento específico a la etiología infecciosa, por lo que los pacientes estables pueden recibir hasta 72 horas de manejo antibiótico para valorar la respuesta y conocer si requerirán o no tratamiento de inducción3.
La meta del tratamiento es destruir las células inmunes que se encargan de respuesta inflamatoria exagerada con un régimen que consiste en dexametasona y etopósido por vía sistémica, y si existe afectación de sistema nervioso central se puede usar metotrexato e hidrocortisona, ambos intratecales. En los protocolos de 2004 se añade ciclosporina3.
El tratamiento se divide en 2 fases8:
Terapia inicial (inducción): Incluye las primeras 8 semanas de tratamiento con las siguientes dosis:
Dexametasona, 10 mg/m2 las semanas 1 y 2, seguido de una disminución de 50% de la dosis cada 2 semanas hasta llegar a 0 en la semana 8.
Etopósido, 150 mg/m2, 2 veces a la semana las semanas 1 y 2, seguido de una dosis semanal de las semanas 3 a 8.
Alemtuzumab, solo si existe contraindicación para el uso de etopósido.
Terapia de continuación: Solo se emplea en casos documentados de enfermedad familiar (principalmente en niños) y en enfermedad no familiar persistente. El esquema comienza en la semana 9 e incluye los siguientes fármacos:
Es importante dar seguimiento a la respuesta al tratamiento con examen físico y estudios de laboratorio (biometría hemática, tiempos de coagulación, fibrinógeno, dímero D, ferritina, creatinina, electrolitos séricos, pruebas de función hepática y en caso de ser necesario análisis de líquido cefalorraquídeo); dependiendo de la condición clínica del paciente estos estudios se realizarán diario (excepto la punción lumbar) en pacientes críticamente enfermos o se pueden realizar más espaciados en pacientes estables5.
La mayoría de los casos en adultos responderán al tratamiento y el trasplante alogénico de médula ósea que está reservado principalmente para niños en los que se documentan mutaciones genéticas, en casos de pacientes con neoplasias hematológicas, pacientes con falta de respuesta a la terapia de inducción o en aquellos con afectación del sistema nervioso central.
En conclusión, el síndrome hemofagocítico es una enfermedad poco sospechada en la práctica clínica ya que sus signos y síntomas son inespecíficos por lo que el diagnóstico suele realizarse de manera tardía. Es importante considerarlo en los pacientes con fiebre y citopenias de etiología indeterminada para brindar un tratamiento oportuno, disminuir las complicaciones y a mortalidad.

