











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Tetrasomy 18p or isochromosome 18p (OMIM; 614290) is a rare chromosomal disorder with a prevalence of 1:140,000-180,000 live births1. At present, more than 200 cases have been described in the literature2,3. Approximately 30% are familial cases and only few cases have been reported as mosaics2. In most cases, the origin of tetrasomy 18p is due to maternal non-disjunction during meiosis II, followed by poor centromere division. Maternal age may play a role in the formation of tetrasomy 18p4. The marker chromosome is an abnormal small chromosome fragment that we can find when performing a karyotype, it has an incidence of 0.14-0.72/1000 births, and many of them correspond to acrocentric chromosomes5. Multiplex ligation-dependent probe amplification (MLPA) is a variation of the multiplex polymerase chain reaction that allows multiple regions of the genome to be amplified at the same time6. The purpose of this study is to report on the MLPA and single nucleotide polymorphism (SNP)-array based molecular analysis of a chromosomal marker in two children that were born with tetrasomy 18p. Interestingly, we observed atopic dermatitis, café-au-lait spots (CALS), and a thyroglossal cyst in one patient. These clinical features have not been previously reported concerning this condition, which could indicate that they are clinical data associated with this syndrome.
Patient 1
A male patient, 3 years and 10 months old, the third child of healthy, non-blood related parents. The mother was 35 years old and the father was 36 years old at the time of birth. Family history was negative for intellectual disability or congenital malformations. No history of prenatal exposure to teratogens or maternal diseases was recorded. After an uneventful pregnancy, the child was born by caesarean section at 40 weeks of gestation with a weight of 3400 g (50th percentile), a height of 49 cm (50th percentile), and a head circumference of 35 cm (50th percentile). Apgar score was 8-9 and did not require special neonatal management. The child's growth and development were delayed, with the child holding its head up at the age of one, being able to remain in seated position at 2 years and it does not recognize receptive or expressive language. The mother reports that her child is very frightened indoors and often gets sick of the airways. From the child's birth to 6 months, he presented atopic dermatitis which has been decreasing.
On physical examination, his weight was 14,000 g (10-25th percentile), height was 104 cm (75th percentile), and head circumference was 49 cm (25-50th percentile). His head is dolichocephalic with prominent forehead, low set ears, depressed nasal bridge with bulbous tip, palpebral fissures slightly downwards, prominent chest, modified single palmar crease on the left hand, bilateral cryptorchidism, five CALS on the abdomen, and thigh and leg more than 1 cm wide (Fig. 1A and B). The cranial tomography showed cerebral cortical atrophy and the ultrasound scan detected a thyroglossal cyst. Due to cryptorchidism, orchidopexy was performed at the age of 2.
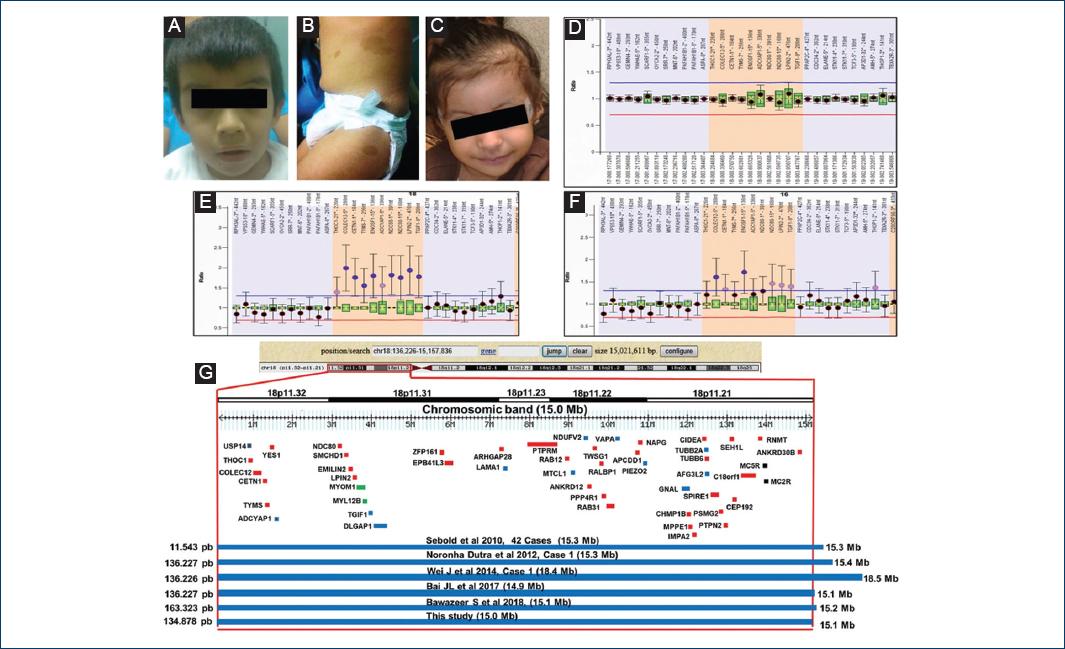
Figure 1 Characteristics of the patients. A-B: patient 1 at the age of 3 years and 5 months. C: patient 2 at the age of 2 years. Café au lait spots in patient. D: multiplex ligation probe amplification (MLPA) of health control. E-F: case 1 and case 2. G: representation of the CytoScan HD result, the figure shows the chromosome 18p11.32p11.21 and the relative position of the genes (USP14, ADCYAP1, DLGAP1, LAMA1, MTCL1, NDUFV2, VAPA, PIEZO2, TUBB2A, AFG3L2, GNAL) involved with the nervous system (blue). The OMIM genes are shown in red. Some affected patients with suppression in the 18p11.32p11.21 region are also shown.
Patient 2
Female patient, 2 years old, the first child of healthy, non-blood related parents. The mother was 36 years old and the father was 33 years old at the time of birth. Family history was negative for intellectual disability or congenital malformations. No history of prenatal exposure to teratogens or maternal diseases was recorded. The mother had two threatened abortions in the first trimester and pre-eclampsia in the last trimester. Obstetric ultrasound showed delayed intrauterine growth. She was born by cesarean section at 37 weeks of gestation with a weight of 2780 g (10th percentile), height of 49 cm (50th percentile), head circumference of 33 cm (10th-25th percentile), and Apgar score of 8-9. The neonatal period was normal. The child's growth and development were delayed, with the child holding its head up at the age of 9 months, being able to remain in seated position at 1 year and 3 months and monosyllable speech at 1 year and 5 months. The child is not able to say complete sentences and does not walk. During physical examination, her weight was 8800 g (percentile < 3), height of 90 cm (90th percentile), cephalic perimeter of 43.5 cm (percentile < 3), microcephaly, ear length measurement within set limits, small palpebral fissures, and convergent strabismus (Fig. 1C). Transfontanellar ultrasound and brain MRI showed frontal cortical atrophy. Renal ultrasound detected right renal hypoplasia.
Materials and methods
The karyotype test was performed in metaphase from cultures of peripheral blood lymphocytes of the patients and their parents with standard methodology for GTG banding (∼500 bands). MLPA reaction was performed using the SALSA MLPA kit P290-B2 and P249 (MRC-Holland, Amsterdam, Netherlands) following supplier instructions. The results were analyzed with the Coffalyser software (MRC-Holland, Amsterdam, Netherlands). The SNP-array analysis was carried out using the CytoScan HD matrix (SNP-array) following the protocol provided by the supplier (Affymetrix, Santa Clara, California, USA). The SNP-array was scanned with the Affymetrix GeneChip Scanner 3000 7G, and the data were analyzed with GTYPE (GeneChip Genotyping Analysis Software, version 1.0.12) to detect aberrations in the number of copies. The resolution of this procedure was estimated at 1.15 kb. The Research Committee of the Institution approved the protocol and the parents gave their informed consent for the participation in the study.
Results
The results of the conventional karyotype test were 47, XY, + mar for case 1 and 47, XX, + mar for case 2. The parents' karyotype tests were normal. The characterization of the marker chromosomes was first analyzed with MLPA. It showed a higher copy number of the 18p THOC1 probe (18p11.32) with the SALSA MLPA kit P290-B2 for both patients, and showed a higher copy number of probes COLEC12, CETN1, TYMS, ENOSF1, ADCYAP1, NDC80, LPIN2, and TGIF1 with the SALSA MLPA kit P249 (Fig. 1D-F). The CytoScan HD array showed a 15 Mb amplification of 18p11.32-p11.21, with 78 RefSeq genes in the genomic variation database, DGV database (http://projects.variation.tcag.ca/variation/hg38/). There are 52 genes listed in the OMIM database (Fig. 1G). The final cytogenetic result for the patients was 46, XY, arr [hg19] 18p11.32-p11.21 (136,226-15,157,836) × 4 dn y 46, XX, arr [hg19] 18p11.32- p11.21 (134,878, −15,149,748) × 4 dn. Parental studies showed normal results.
Discussion
In the present study, two children are described with craniofacial dysmorphism, psychomotor retardation, significant language delay, short stature, and cortical atrophy due to tetrasomy 18p. Both patients presented marker chromosomes identified by GTG banding; MLPA analysis showed the amplification of the short arm of chromosome 18 and the SNP-array analysis delimited the tetra-amplification of 18p.
At present, only few cases with tetrasomy 18p have been analyzed with MLPA and SNP-array1,3,7-10. Previous analysis of a group of patients with tetrasomy 18p identified that the first ten characteristics in these patients were developmental delay, intellectual disability, abnormal muscle tone, feeding difficulties, variants in brain MRI, microcephaly, strabismus, cryptorchidism, scoliosis/kyphosis, constipation, and growth retardation1. Heart, neural tube closure, and palatal defects were also observed to a lesser extent. Characteristics such as retrognathia, articular contraction of the limbs, and hypoplastic nails have also been described. Microcephaly, oval face, strabismus, small nose, low set ears, constipation, anomalies in the left foot, cryptorchidism, tendency to aggression, and autism were observed as symptoms in a 5-year-old boy with tetrasomy 18p7. Our patient 2 showed similar characteristics to those reported; however, patient 1 showed atopic dermatitis, CALS and a thyroglossal cyst. This data had not been previously reported, which we deemed likely to be part of the clinical condition.
In the case of molecular analysis, the amplification of 15.0 Mb at 18p included 52 genes listed in the OMIM database, 12 of which are related to the nervous system and may have some impact on the phenotypic characteristics of patients. The MC2R gene is expressed in the skin and can play a role in the pathophysiology of the skin11, such as the CALS observed in our patient. The product of the USP14 gene is involved in the degradation of proteasome substrates, can regulate synaptic activity and is related to Huntington's disease, affective disorders, and schizophrenia12. The PACAP gene is expressed in lateral ventricles, dentate line and hypothalamus producing the proliferation of nervous system cells, and has been associated with changes in sensitivity, learning and olfactory aspects13. The TGIF gene is related to the development of brain disease and has been observed to be expressed in the cortex of mice14. The DLGAP gene is expressed in synaptic sites of hippocampal dendritic spine and cerebral cortex of mice15. The MTCL1 gene is related to the packaging and stabilization of microtubules in neurons16. The LAMA1 gene is related to intellectual disability and speech delay, as well as cysts and cerebellar atrophy, myopia, retinal dystrophy, and abnormal eye movement in Poretti-Boltshauser syndrome17. The NDUFV2 gene has been associated with encephalopathy, hypertrophic cardiomyopathy, and truncal hypotonia18. The VAPA gene is highly expressed in the brains of mice with unknown function19. The PIEZO2 gene has a role in electrical conduction that is mechanically activated in somatosensory neurons in the dorsal root ganglia; its defects have been related to Marden-Walker syndrome and distal arthrogryposis20. The GNAL gene is expressed in the basal ganglia, caudal nuclei, and in the amygdala and is associated with dystonia21. The TUBB2A gene has been associated with the phenotype of cerebral cortical dysplasia and other malformations while the AFG3L2 gene is related to cerebellar ataxia, dystonia, myoclonic epilepsy, oculomotor apraxia, and decreased ambulation22,23. It is worth mentioning that it is important to carry out more molecular studies of the nervous system genes to determine their participation in the phenotype of patients with tetrasomy 18p.
Conclusion
The present study describes two patients with tetrasomy 18p by MLPA and SNP-array analysis, with a previous diagnosis of chromosomal marker by conventional karyotype test. We observed new previously unreported characteristics in one of the patients with tetrasomy 18p. We also emphasize the importance of easy and fast molecular analysis of the marker chromosomes by MLPA.

