











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Antecedentes
La enfermedad granulomatosa crónica (EGC) es una inmunodeficiencia primaria causada por deterioro funcional del complejo enzimático nicotinamida adenina dinucleótido fosfato (NADPH) oxidasa. Esta enfermedad se caracteriza por infecciones severas y recurrentes, hiperinflamación y autoinmunidad. El complejo NADPH oxidasa se compone de proteínas citosólicas y de membrana que se unen para la activación de fagocitos para producir superóxido. A partir del superóxido se generan otros radicales libres de oxigeno (RLO) esenciales para destruir bacterias, hongos y micobacterias. Las mutaciones en cualquiera de los genes que codifican para las cinco subunidades estructurales de la NADPH oxidasa (gp91phox, p22phox, p47phox, p67phox, p40phox) y recientemente la proteína chaperona EROS, dan como resultado una producción defectuosa de RLO y el síndrome de la EGC.1
Epidemiología de EGC
En la mayoría de los casos, la EGC se diagnostica en los primeros tres años de vida. La incidencia estimada a nivel mundial es de uno en aproximadamente 12 000 a 250 000 recién nacidos vivos. En México se desconoce la incidencia. Las cohortes estadounidenses y europeas con 368 y 429 pacientes, respectivamente, son las más grandes reportadas hasta la fecha.2,3
El defecto en gp91phox se hereda de forma ligada al cromosoma X (LX), mientras que los defectos en las proteínas p22phox, p47phox, p67phox, p40phox y CYBC1/EROS se heredan de forma autosómica recesiva (AR). A nivel internacional, según el tipo de herencia, la EGC LX es más frecuente, con 65 a 70 %, sin embargo, la herencia AR de EGC se ha reportado más frecuentemente en regiones del mundo con tasas altas de consanguinidad, como en los países árabes, Israel e India (cuadro 1).3,4,5,6
Cuadro 1 Distribución de enfermedad granulomatosa crónica según el tipo de herencia o subunidad afectada de la NADPH oxidasa, en diferentes cohortes internacionales
| País/región | Autor | Total pacientes (n) | EGC LX (gp91phox) | AR | p47phox | p67phox | p40phox | p22phox |
| Estados Unidos | Winkelsteinet al.4 | 368 | 259 | 81 | 45 | 10 | 0 | 7 |
| Túnez | El Kareset al.55 | 15* | 0 | 15 | 5 | 5 | 0 | 3 |
| Italia | Martireet al.56 | 60** | 39 | 6 | 5 | 0 | 0 | 1 |
| Europa | Van der Berget al.5 | 429 | 290 | 139 | 69 | 11 | 0 | 22 |
| India | Rawatet al.3 | 17 | 7 | 10 | 7 | — | — | — |
| Turquía | Kokeret al.41 | 89 | 34 | 50 | 17 | 13 | 0 | 20 |
| Estados Unidos | Bartolettoet al.57 | 27 | 19 | 8 | 2 | 2 | — | — |
| Latinoamérica | De Oliveira jr.et al.15 | 71 | 53 | 18 | 16 | 0 | 0 | 2 |
| Israel | Wolachet al.6 | 84 | 32 | 52 | 26 | 16 | 0 | 10 |
*Dos pacientes se reportaron como desconocidos. **En 25 pacientes la herencia no fue determinada. NADPH = nicotinamida adenina dinucleótido fosfato. AR = autosómica recesiva,
En la cohorte estadounidense de 368 pacientes, los niños con EGC LX tuvieron tasas significativamente más altas de infecciones graves en comparación con los niños con EGC AR. La mortalidad en la EGC LX y la EGC AR fue de 21.2 y 8.6 %, respectivamente (p < 0.02). En la cohorte europea de 439 pacientes, la EGC AR se diagnosticó en edades más tardías y la supervivencia fue significativamente mayor (49.6 años), comparada con la EGC LX (37.8 años).4,5
En la cohorte israelí de 84 pacientes, 52 (62 %) fueron diagnosticados con herencia AR y 32 (38 %) con herencia LX. Se detectó consanguinidad en 64 % de familias con herencia AR. El espectro clínico de los 84 pacientes varió de leve a severo. El tipo de EGC AR fue generalmente más leve, además, se encontró en este grupo una mayor producción residual de RLO, lo cual se correlacionó con una expresión clínica más leve, un mejor pronóstico y una mayor supervivencia.6
Ha habido algunos informes que sugieren que el pronóstico en pacientes con EGC está determinado principalmente por la cantidad de actividad residual de la NADPH oxidasa, independientemente del modo de herencia y esto está influido por el tipo de mutación en el gen afectado. Por lo tanto, la actividad residual de la NADPH oxidasa puede guiar el modo de terapia en un paciente. En general, con el reconocimiento temprano de la enfermedad, la profilaxis antimicrobiana a largo plazo y el tratamiento de pacientes seleccionados con trasplante de células progenitoras hematopoyéticas, la supervivencia de los pacientes con EGC ha mejorado a lo largo del tiempo.4,7
Inmunopatología
La enzima NADPH oxidasa se expresa en neutrófilos, monocitos, macrófagos, eosinófilos y células dendríticas, así como linfocitos B y T. La expresión de las subunidades de la NADPH oxidasa y su actividad enzimática es mayor en los neutrófilos y en los eosinófilos. La mayoría de la actividad catalítica de los fagocitos, como los neutrófilos, se atribuye a la capacidad para producir grandes cantidades de RLO por el complejo NADPH oxidasa. Este complejo multimérico cataliza la transferencia de los electrones de oxígeno, generando superóxido (O2 -), precursor de múltiples RLO con funciones microbicidas e inmunorreguladoras. El superóxido es un compuesto antimicrobiano relativamente débil y tiene una vida media muy corta, sin embargo, el O2 - sirve como precursor de otros potentes RLO, cruciales para eliminar diferentes especies de bacterias, micobacterias y hongos.8,9
Las mutaciones AR o LX en los genes del complejo NADPH oxidasa resultan en disminución o ausencia de RLO y dan como consecuencia la patología denominada EGC clásica. La EGC clásica se caracteriza por infecciones recurrentes y severas por bacterias, hongos y micobacterias, además de manifestaciones asociadas con una inflamación aberrante. La NADPH oxidasa está implicada en la erradicación de ciertos agentes infecciosos, en la digestión de antígenos proteicos y en procesos celulares regulados por redox que limitan la inflamación.10
Las subunidades que activan al complejo incluyen el flavocitocromo b, un heterodímero compuesto por gp91phox (NOX2) y p22phox, el cual está localizado en las membranas citoplasmáticas y del fagosoma, y cuatro subunidades regulatorias que se desplazan del citosol hacia la membrana para realizar la activación celular: p47phox, p67phox, p40phox y la forma de Rac unida a GTP (figura 1 y cuadro 2). La fosforilación de p47phox y la formación de Rac-GTP son dos eventos clave en el ensamblaje del complejo de oxidasa activa en la membrana. La subunidad p67phox es un cofactor catalítico involucrado en la transferencia inicial de electrones desde NADPH al flavocitocromo b, mientras que las otras subunidades cumplen varios papeles en la regulación del ensamblaje y la actividad del complejo NADPH oxidasa.2,11
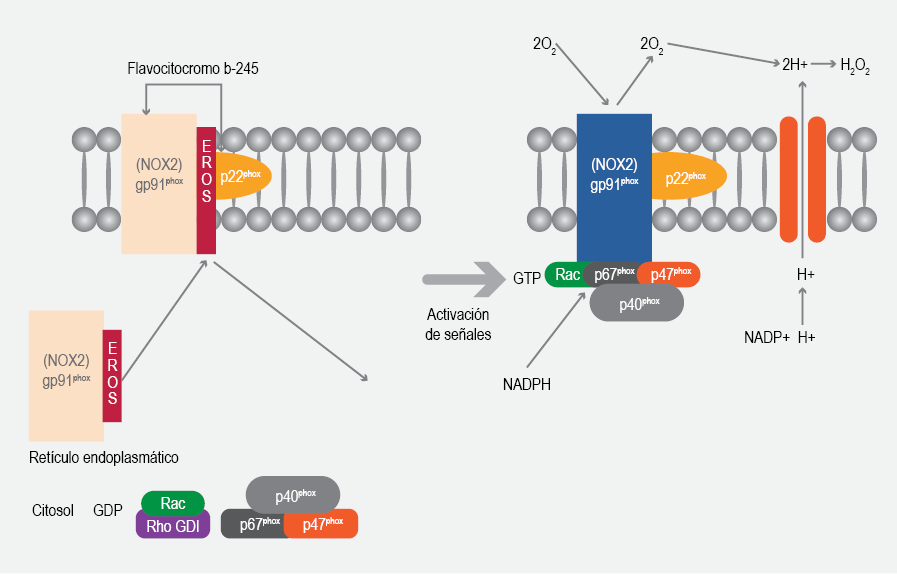
Figura 1 Representación esquemática de las formas inactivas y activas del complejo NADPH oxidasa. El complejo consta de seis subunidades. NOX2 (gp91phox) y p22phox se asocian para formar un heterodímero (flavocitocromo b-245) unido a la membrana plasmática, tanto en la forma inactiva como en la activa. La proteína transmembranal EROS, que inicialmente se localiza en el retículo endosplasmático, actúa como una enzima chaperona para la dimerización de gp91phox y p22phox (flavocitocromo b-245), sin embargo, aún no se conoce del todo su localización y función en relación con el complejo NADPH oxidasa. En reposo, p47phox, p67phox, p40phox y la proteína G Rac se encuentran en el citosol. En la forma activa, las subunidades citosólicas se asocian con el heterodímero NOX2 (gp91phox)/p22phox unido a la membrana. La NADPH oxidasa activa genera superóxido (O2 -), transfiriendo electrones de NADPH dentro de la célula a través de la membrana y acoplándolos al oxígeno molecular.
Cuadro 2 Genes y proteínas afectadas en la enfermedad granulomatosa crónica
| Gen mutado | Proteína afectada | Localización cromosomal | Tamaño en kb | Exones (n) | Tipo de herencia |
| CYBB | NOX2 o gp91phox | Xp21.1 | 30 | 13 | Ligada al cromosoma X |
| CYBA | p22phox | 16q24 | 8.5 | 6 | Autosómica recesiva |
| NCF1 | p47phox | 7q11.23 | 15 | 11 | Autosómica recesiva |
| NCF2 | p67phox | 1q25 | 40 | 16 | Autosómica recesiva |
| NCF4 | p40phox | 22q13.1 | 18 | 10 | Autosómica recesiva |
| CYBC1 | CYBC1 o EROS | 17q25.3 | 11 | 9 | Autosómica recesiva |
Recientemente se ha descrito una cohorte de 24 pacientes con deficiencia de p40phox, con niveles más altos de producción de RLO y predominio de las manifestaciones inflamatorias sobre las infecciones, que se diferencian de la forma clásica de EGC. Los RLO producidos por los neutrófilos pueden contribuir a la depuración de células apoptóticas o residuos celulares, esta deficiencia podría favorecer la inflamación a través de diferentes vías como la esferocitosis aberrante.12
La proteína transmembranal EROS codificada por el gen CYBC1 es otra subunidad esencial para la formación del complejo NADPH oxidasa, ya que actúa como una enzima chaperona en el proceso de dimerización de gp91phox y p22phox (citocromo b-245). En individuos con deficiencia de esta proteína, los niveles de RLO son bajos (y no ausentes), como en la EGC por deficiencia de p47phox.13
Cuadro clínico
El cuadro clínico de la enfermedad granulomatosa crónica (EGC) se distingue por infecciones recurrentes causadas por diversos agentes etiológicos (bacterias, hongos y micobacterias) y un estado inflamatorio que resulta en la formación de granulomas.14,15
A la fecha, diversos estudios reportan que los órganos más frecuentemente afectados en la EGC, tanto por los agentes infecciosos como por los granulomas, incluyen los pulmones, ganglios linfáticos, hígado y piel (cuadro 3).4
Cuadro 3 Cohortes internacionales de enfermedad granulomatosa crónica, con las manifestaciones clínicas más frecuentes
| Cohorte | Principales síntomas | % |
| América Latina 71 pacientes (2015) | Neumonía recurrente | 76.8 |
| Linfadenopatía | 59.4 | |
| Granulomas | 49.3 | |
| Europa 429 pacientes (2009) | Involucro pulmonar (neumonía/abscesos/granuloma) | 66 |
| Afección cutánea (abscesos/granulomas) | 53 | |
| Linfadenitis | 50 | |
| China 169 pacientes (2018) | Fiebre recurrente | 36.8 |
| Infección pulmonar (neumonía) | 31.6 | |
| Impétigo | 16.8 | |
| Israel 84 pacientes (2017) | Infecciones (neumonía, adenitis supurativa, abscesos cutáneos/subcutáneos | Sin especificar |
| Granulomas (hígado/pulmón) | 66 | |
| Abscesos hepáticos | 39 | |
| Estados Unidos 368 pacientes (2000) | Neumonía | 79 |
| Abscesos (subcutáneo, hepático, pulmonar) | 68 | |
| Adenitis supurativa | 53 |
Las series más grandes de pacientes con EGC, en su mayoría documentan la neumonía recurrente como la principal condición clínica de estos pacientes, en algunas ocasiones asociadas con derrame pleural. Otras manifestaciones reportadas son linfadenopatías, granulomas, infecciones cutáneas, diarrea crónica, otitis, sepsis, abscesos (hígado, pulmón, área perianal, cerebro y riñón), infecciones de vías urinarias recurrentes y osteomielitis (cuadro 3).5,6 Por otra parte, entre los agentes etiológicos más frecuentes se encuentran Staphylococcus aureus, Escherichia coli, Aspergillus spp., Serratia marcescens, Candida spp., Klebsiella spp., Salmonella y Nocardia spp. (cuadro 4).15 La infección por el complejo Burkholderia cepacia es común en los pacientes con EGC y ocasionalmente pueden causar sepsis. Las infecciones bacterianas inusuales consideradas como virtualmente patognomónicas para EGC son Granulibacter bethesdensis, Chromobacterium violaceum y Francisella philomiragia.16,17,18
Cuadro 4 Cohortes internacionales de enfermedad granulomatosa crónica, con los agentes infecciosos más frecuentes
| Cohorte | Agente infeccioso |
| América Latina 71 pacientes (2015) | BCG |
| Staphylococcus aureus | |
| Aspergillus spp. | |
| Europa 429 pacientes (2009) | Staphylococcus aureus |
| Aspergillus spp. | |
| Salmonella | |
| China (pulmón) 169 pacientes (2018) | Staphylococcus aureus |
| Klebsiella pneumoniae | |
| Moraxella catarrhalis | |
| Israel 84 pacientes (2017) | Staphylococcus aureus |
| Aspergillus spp. | |
| Serratia marcescens | |
| Estados Unidos 368 pacientes (2000) | Aspergillus |
| Staphylococcus spp. | |
| Serratia spp. |
Los porcentajes de frecuencia no fueron incluidos por las diversas formas de clasificación en cada estudio.
Las infecciones fúngicas invasivas (en las que la EGC tiene la mayor prevalencia en el grupo de las inmunodeficiencias primarias) se mantienen como la principal causa de mortalidad en estos pacientes. Se manifiestan clínicamente como neumonía, abscesos/granulomas, sepsis y linfadenitis. Se reporta un mayor riesgo de incidencia en las primeras dos décadas de la vida y 30 % de probabilidad de padecerla a lo largo de la misma.6,19,20
Las enfermedades micobacterianas en pacientes con EGC están principalmente limitadas a infecciones por BCG y tuberculosis a nivel local y regional.21 En una cohorte en la que se incluyeron 71 pacientes que habían tenido alguna infección micobacteriana y de los cuales 70 habían recibido la vacuna BCG, 63 % presentó BCGitis y 37 % BCGosis. Los sitios más frecuentemente afectados por las infecciones micobacterianas fueron los ganglios linfáticos regionales, ganglios linfáticos sistémicos, pulmón, hígado, tejidos blandos, meninges, piel, hueso y bazo. Treinta y un pacientes (44 %) desarrollaron tuberculosis. Los sitios más frecuentemente afectados fueron ganglios linfáticos periféricos (26 %), huesos y meninges (3.2 %).10
Otras manifestaciones
Una de las principales características de la EGC es la detención del crecimiento, con falta de ganancia de peso. Los pacientes que llegan a la adolescencia presentan peso y talla por debajo de los esperados.22
El estado proinflamatorio, que caracteriza a esta enfermedad, se desencadena principalmente en respuesta a la presencia de agentes infecciosos y a largo plazo puede afectar diversos aparatos y sistemas del organismo.
Se ha encontrado que las manifestaciones crónicas más frecuentes son anemia por infección crónica (65 %), linfadenopatía (52 %), hepatoesplenomegalia (25 %) y alteraciones gastrointestinales (25 %). A nivel pulmonar son más frecuentes bronquiectasias, bronquiolitis obliterativa y fibrosis crónica (figura 2). Por otra parte, las manifestaciones cutáneas incluyen fotosensibilidad, lesiones granulomatosas y vasculitis.6,23
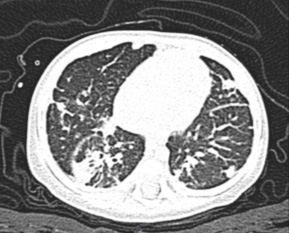
Figura 2 Complicaciones pulmonares crónicas en enfermedad granulomatosa crónica ligada al cromosoma X
La formación de granulomas y el estado inflamatorio contribuyen a la morbilidad y a la presencia de múltiples síntomas en este padecimiento, con lo que se afecta principalmente los sistemas genitourinario y gastrointestinal. Las manifestaciones de los tractos genitourinario y gastrointestinal incluyen obstrucción uretral por granulomas e infecciones de vías urinarias recurrentes y, con menor frecuencia, pseudotumores vesicales y cistitis eosinofílica, obstrucción intestinal, granulomas (esofágicos, yeyunales, ileales, cecales, rectales y perirrectales), dolor abdominal, diarrea, apendicitis, pancreatitis, estenosis y fístulas. El estado inflamatorio crónico puede superponerse incluso con síndrome hemofagocítico.5,14,22,24,25,26,27
A nivel hepático, la EGC puede presentar abscesos (se observan en más de 35 % de los pacientes), enzimas de función hepática elevadas, hepatitis, hepatoesplenomegalia, hiperplasia nodular regenerativa e hipertensión portal.28,29
Las manifestaciones oftalmológicas incluyen lesiones coriorretinianas, granulomatosas, queratitis y uveítis. La cavidad oral también puede ser afectada y clínicamente estos pacientes pueden presentar gingivitis, estomatitis, aftas e hipertrofia gingival. De igual forma se ha descrito la pérdida temprana de dientes, principalmente en las formas autosómicas recesivas.14,30,31
Los desórdenes de etiología autoinmune tales como púrpura trombocitopénica, artritis idiopática juvenil, miastenia gravis, nefropatía por IgA y síndrome de anticuerpos antifosfolípidos tienen mayor prevalencia en la EGC. Se han presentado hasta 50 % de los individuos afectados e, incluso, pueden ser la forma de presentación inicial de la enfermedad.4,22,32
A pesar de que hay reportes aislados de pacientes con EGC y neoplasias, en las series más grandes no se ha registrado asociación clara entre la EGC y mayor riesgo de desarrollo de neoplasias malignas.4,5
Una variedad distinta de EGC no “clásica” es la causada por deficiencia de p40phox; las manifestaciones autoinmunes y autoinflamatorias predominan sobre las infecciones graves e invasivas. El inicio de las manifestaciones es más tardío, con promedio de inicio de seis años de edad. Los pacientes pueden presentar lesiones similares a lupus o lupus discoide, úlceras orales, periodontitis, gingivitis, abscesos/granulomas gastrointestinales, esofagitis, gastritis, enfermedad de Crohn o abscesos/fístulas perianales. Las infecciones son menos invasivas, incluyendo la aspergilosis.12
Recientemente se describió una nueva forma de EGC secundaria a mutación en el gen CYBC1 que codifica para la proteína EROS. En esta nueva forma, las manifestaciones clínicas distintivas son colitis difusa, infecciones por Lysteria monocitogenes, Legionella, Salmonella typhimurium y Candida septicemia. Una característica importante de esta variedad de EGC es el desarrollo tardío de las manifestaciones clínicas, con un promedio de inicio de la enfermedad de ocho años de edad.13,33
Diagnóstico
El diagnóstico de EGC se realiza con pruebas de laboratorio que se basan en la medición directa de los RLO, superóxido (O2 −) y peróxido de hidrógeno (H2O2), ambos producidos por los neutrófilos a través del complejo enzimático NADPH oxidasa.
Las técnicas que miden la producción de superóxido son la reducción de ferrocitocromo C, isoluminol, nitrato de bis-N-metilacridinio (lucigenin) y el nitroazul de tetrazolio (NBT). El peróxido de oxígeno se cuantifica con otras técnicas que utilizan citometría de flujo; su uso se ha incrementado con el tiempo, ya que permiten detectar el patrón de herencia de la enfermedad y son ensayos más objetivos y sensibles. En estas técnicas, los neutrófilos in vitro se ponen en contacto con forbol-12 miristato-13 acetato (PMA) u otro estímulo (partículas de zimosan tratadas con suero, N-formil-metionina-leucil-fenilalanina, Staphylococcus aureus o Escherichia coli) y se agrega un compuesto no fluorescente que al interaccionar con los RLO se transforma en un compuesto fluorescente, que se mide en el citómetro de flujo. La EGC por deficiencia en p40phox se detecta con estímulos particulados. Los fluorocromos utilizados son la 2,7 diclorohidrofluoresceína, 10-acetil-3,7-dihidroxifenoxazina (resorufina), 5-amino-2,3-dihidro-1,4-ftalazindiona (luminol), hidroetidina, 6G dihidrorodamina y la 1,2,3 dihidrorodamina (DHR). Esta última es la más usada para el diagnóstico de EGC por su mayor sensibilidad.11,27,34,35,36
Diagnóstico del patrón de herencia de la EGC
La técnica de 1,2,3 DHR permite diferenciar entre las formas recesiva LX o AR. En la enfermedad LX, los varones son afectados y las mujeres se comportan como portadoras. En las formas AR, ambos sexos pueden verse afectados y ambos padres son portadores obligados. En las portadoras de EGC-LX, debido al fenómeno de lionización, existen dos poblaciones de neutrófilos, una con producción normal de RLO y otra sin producción de estos. A través de la técnica de 1,2,3 DHR podemos observar en el histograma ambas poblaciones (figura 3). Si hay un varón con EGC por DHR y su madre tiene un patrón bimodal, puede asegurarse que se trata de una EGC-LX; sin embargo, si tenemos un varón con ECG por DHR y su madre no tiene un patrón bimodal, no podemos determinar el patrón de herencia, ya que puede tratarse de EGC-LX con una mutación de novo o una EGC autosómica recesiva. Existe una variante para las portadoras conocida como inactivación aleatoria del X, en donde, por algún factor aún desconocido, existe inactivación preferencial del gen CYBB wild type y transcripción solo del gen CYBBcon la mutación, lo cual explica la EGC-LX en una mujer (figura 3).36,37,38
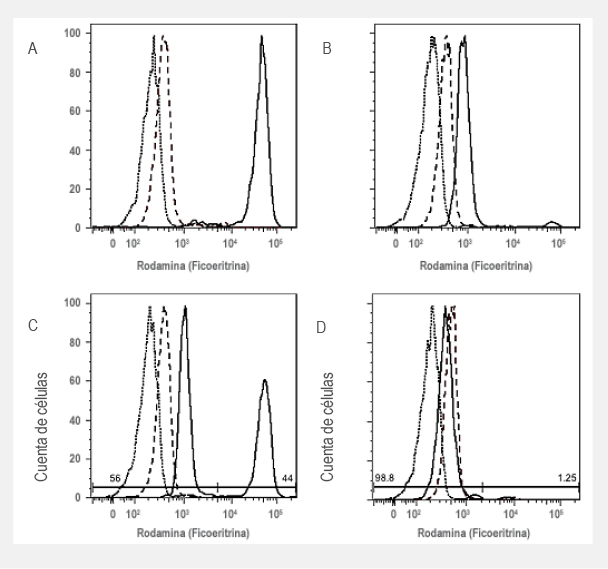
Figura 3 Análisis de la producción de peróxido de hidrógeno (H2O2) por neutrófilos en sangre periférica, mediante la técnica 1,2,3 dihidrorodamina (DHR). Los histogramas de muestras de sangre sin estimulación se muestran en línea punteada, sin estimulación de forbol-12-miristato-13-acetato (PMA) en línea discontinua y con estimulación de PMA en línea continua. A) Control sano con desplazamiento unimodal en la intensidad media de fluorescencia hacia la derecha después de la estimulación con PMA. B) Paciente con enfermedad granulomatosa crónica, quien presenta mínima respuesta por parte de los neutrófilos para la producción de radicales libres posterior a la estimulación con PMA. C) Mujer portadora de enfermedad granulomatosa crónica quien presenta dos poblaciones diferentes de neutrófilos posterior a la estimulación con PMA, una con producción normal de H2O2 (45.3 %) y otra con producción anormal (54.7 %). D) Portadora de enfermedad granulomatosa crónica con inactivación aleatoria del cromosoma X sano, quien presenta una producción normal de H2O2 únicamente en 4.11 % de sus neutrófilos después de la estimulación con PMA.
Se debe extender una invitación para detectar otras posibles portadoras en la familia como las hermanas, abuela, tías, primas y sobrinas por vía materna del paciente. A las portadoras de EGC-LX se les debe ofrecer asesoramiento genético y advertir sobre los diversos síntomas que pueden presentar.39
Expresión de las subunidades de la enzima NADPH oxidasa
Se puede detectar de forma individual cada una de las diferentes subunidades de la NADPH oxidasa a través de una tinción intracelular, para ello se utilizan anticuerpos monoclonales específicos (primarios) y estos a su vez son marcados con un anticuerpo secundario. En un paciente con EGC cuya madre es identificada como portadora por técnica de 1,2,3 DHR se evalúa la expresión de gp91phox , tanto en el paciente como en la madre; en la mayoría de los casos de EGC-LX se observa ausencia de la proteína en los pacientes y en las portadoras se presentan dos poblaciones, una con expresión de la proteína normal y otra sin expresión. En los pacientes varones cuyas madres no son portadoras de la enfermedad y en las mujeres con EGC se debe realizar la medición de gp91phox, p22phox, p67phox y p47phox (junto con un control sano), una de las cuatro subunidades estará ausente o disminuida en comparación con el control sano; este resultado orienta al gen afectado, que debe ser secuenciado.11,40
Análisis de mutación
La mutación causante de la enfermedad debe ser determinada mediante secuenciación Sanger de ADN genómico en cada paciente. Con base en el resultado debe otorgarse el asesoramiento genético. Las portadoras de la enfermedad sin síntomas clínicos pueden ser diagnosticadas confiablemente mediante análisis de mutación del gen CYBB. Asimismo, cuando los pacientes son trasplantados con células progenitoras hematopoyéticas de un miembro de la familia es importante saber que este donante no está portando la mutación y si el diagnóstico prenatal o la terapia génica es una opción, esta información es imprescindible.
El ADN genómico y el ARN se pueden extraer de células mononucleares de sangre periférica o de neutrófilos en sangre total. Los pares de cebadores intrónicos que flanquean cada uno de los exones del gen afectado son amplificados mediante PCR y se realiza secuenciación Sanger de ADN genómico.41,42
Tratamiento
Profilaxis
El tratamiento de la EGC es primordialmente con antibióticos y antifúngicos profilácticos de por vida. La terapia profiláctica se basa en dosis diarias de trimetoprim/sulfametoxazol (TMP/SMX) e itraconazol, que reducen la frecuencia de las principales infecciones bacterianas y fúngicas, respectivamente. La profilaxis medicamentosa ha contribuido a mejorar la supervivencia de los pacientes; sin la profilaxis, el promedio de episodios infecciosos potencialmente mortales era de 10 meses y con su uso, la tasa de infección se ha reducido a un episodio cada tres a cuatro años.43,44
La profilaxis con TMP/SMX suele administrarse a 5 mg/kg/día (con base en TMP) hasta 320 mg/día. Los efectos adversos más frecuentes pueden interferir con la adherencia a la medicación e incluyen síntomas gastrointestinales (dolor abdominal, diarrea y, rara vez, pancreatitis) y dermatológicos, (erupciones cutáneas, fotosensibilidad y, rara vez, síndrome de Stevens-Johnson). La reacción más común a TMP/SMX es una erupción morbiliforme y fiebre asociada que ocurre de siete a 12 días después del inicio del tratamiento. La dicloxacilina y la ciprofloxacina son opciones para pacientes con alergia al sulfametoxazol o deficiencia de G6PD.1,44,45
El itraconazol tradicionalmente ha sido el azol de elección, la dosis recomendada es de 5 a 10 mg/kg/día hasta 200 mg diarios; los principales efectos adversos son elevación de transaminasas, cefalea, náuseas y vómitos. Debido a la intolerancia ocasional a este medicamento y a la aparición de organismos resistentes, el uso de voriconazol y posaconazol ha aumentado.1,44
Tratamiento de infecciones
Cada episodio infeccioso debe considerarse potencialmente peligroso, por lo que se debe hacer todo lo posible para identificar el microorganismo responsable mediante la realización de pruebas no invasivas e invasivas. La terapia empírica inicial debe incluir al menos dos antibióticos contra las bacterias grampositivas y gramnegativas. De no responder dentro de las 48 horas, pueden ser necesarios cambios empíricos en la cobertura de antibióticos antes de la identificación definitiva del patógeno. El tratamiento debe continuarse durante semanas o meses, incluso con una mejora significativa en el índice inflamatorio y la condición clínica de los pacientes, para erradicar la infección por completo.43,44
En los pacientes con síntomas pulmonares o fiebre de origen incierto, la terapia antifúngica debe iniciarse como parte de la terapia empírica inicial. La aspergilosis es la causa principal de infección fúngica invasiva en pacientes con EGC; 26 a 43 % de los pacientes experimenta al menos una infección micótica invasiva en su vida, especialmente neumonía o abscesos cerebrales. Es muy importante resaltar que las pruebas serológicas de Aspergillus y el lavado alveolar bronquial tienen una sensibilidad baja en pacientes con EGC y no deben considerarse como pilar diagnóstico pues solo se retardaría el inicio del tratamiento antifúngico.1,44
El antifúngico recomendado de primera línea es el voriconazol (9 mg/kg/dosis en < 40 kg y 200 mg/dosis > 40 kg cada 12 horas), sin embargo, su uso prolongado se ha asociado con dermatotoxicidad, incluyendo la fotodermatitis y una mayor susceptibilidad a carcinoma de células escamosas y melanoma. Por lo tanto, el voriconazol se debe usar conservadoramente durante seis a nueve meses, especialmente en pacientes con factores de riesgo de cáncer de piel. Cuando las infecciones son resistentes al voriconazol o cuando hay intolerancia a este, la anfotericina B liposomal intravenosa, el posaconazol y la caspofungina han demostrado ser eficaces.43
Los corticosteroides anteriormente no se recomendaban para el tratamiento de EGC; en la actualidad, hay varios informes sobre el uso exitoso de esteroides para el tratamiento de complicaciones inflamatorias e infecciones agudas (principalmente en abscesos hepáticos e infecciones respiratorias) junto con el tratamiento antimicrobiano.25,26,27
Interferón gamma recombinante
El interferón gamma IFN-γ recombinante se ha usado de forma profiláctica en los pacientes con EGC como inmunomodulador desde finales de la década de 1980, ya que ha demostrado estimular el estallido respiratorio, independientemente del defecto genético, y aumentar la actividad bactericida de los fagocitos. Al inyectarse por vía subcutánea tres veces por semana ha reducido la tasa de infección grave en pacientes con EGC en 67 %, así como la frecuencia y duración de las hospitalizaciones.44,46
No se ha encontrado ningún efecto adverso potencialmente mortal relacionado con la terapia con interferón gamma recombinante, sin embargo, puede presentarse fiebre, fatiga, mialgia, rash cutáneo, cefalea y dolor abdominal. La dosis recomendada es de 1.5 μg/kg en pacientes con superficie corporal (SC) ≤ 0.5 m2 y 50 μg/m2 SC en pacientes con > 0.5 m2 SC, vía subcutánea tres veces por semana. Un estudio reciente demostró que una titulación gradual de la dosis reduce la probabilidad de estos efectos adversos, la dosis gradual recomendada es de 15 μg/m2 SC tres veces por semana durante la semana 1, 30 μg/m2 SC tres veces por semana durante la semana 2, seguida de la dosis completa de 50 μg/m2 SC tres veces por semana durante la semana 3.46,47,48
El IFN-γ recombinante debe suspenderse en casos de infección y fiebre. La fiebre puede presentarse como un efecto adverso y puede dificultar la evaluación de la respuesta a los antimicrobianos.
El mecanismo preciso por el cual el IFN-γ recombinante mejora la defensa del huésped en pacientes con EGC sigue sin estar claro; anteriormente se pensaba que revertía el defecto subyacente en la enfermedad, sin embargo, esta teoría ya ha sido descartada. No obstante, la disminución de la tasa de infecciones, la mejora de la supervivencia y la falta de toxicidad significativa sugieren que el tratamiento con IFN-γ recombinante para la EGC sigue estando bien fundamentado.47
Trasplante de células progenitoras hematopoyéticas (TCPH)
El único procedimiento curativo para la EGC disponible actualmente en México es el trasplante de células madre hematopoyéticas (TCPH), que puede revertir las complicaciones infecciosas y las inflamatorias de la enfermedad. Existe una continua controversia sobre las indicaciones y el momento óptimo para el TCPH. La recomendación actual es priorizar el trasplante en los pacientes con ausencia de actividad de la NADPH oxidasa (producción residual ausente), con una o más infecciones potencialmente mortales, falta de apego a la profilaxis antimicrobiana o con inflamación que mejora solo con esteroides.43
Diversos centros hospitalarios han reportado su experiencia en el TCPH. En estos estudios se describen los diversos regímenes empleados, con una supervivencia global de hasta 93 % mediante acondicionamiento mieloablativo o esquemas de acondicionamiento de intensidad reducida. Es importante mencionar que alcanzar la dosis de mieloablación óptima para generar espacio en los nichos receptores e inmunosuprimir al huésped, sin tener sus efectos adversos como enfermedad de injerto contra huésped e infecciones fatales, es todo un reto. Asimismo, el proceso de TCPH en sí mismo tiene limitaciones como disponibilidad de donantes, riesgos asociados con el esquema de acondicionamiento y las complicaciones derivadas de la interacción inmune entre el huésped y el donante.43,49,50,51,52
El estado del injerto en EGC se puede evaluar con la determinación de quimerismo, sin embargo, otras técnicas útiles son la comparación de la 1,2,3 DHR (evalúa el estallido respiratorio) y la expresión positiva de la proteína gp91phox en neutrófilos o monocitos, antes y después del trasplante. Ambas muestran la presencia de una población celular (receptor 100 % o donador 100 %) o dos poblaciones celulares (porcentaje del paciente y porcentaje del donador). En EGC se requiere al menos 10 % de las células del donador para que el paciente esté libre de manifestaciones clínicas. Otras ventajas de estas pruebas son su simplicidad, rapidez y costo-efectividad.53
Terapia génica
La terapia génica para células hematopoyéticas representa una alternativa atractiva al TCPH para pacientes con EGC sin un donante compatible, sin embargo, este tratamiento solo es realizado en Italia, Reino Unido, Francia y Estados Unidos. El procedimiento se realiza ex vivo mediante la corrección del defecto genético en células extraídas del paciente, cultivadas y modificadas genéticamente en el laboratorio. Al dividirse estas células, transmiten el transgen (gen insertado en virus modificado) a sus células hijas. Al paciente solo se le trasfunden las poblaciones celulares en las que se ha comprobado la integración y funcionamiento correcto del transgén.51,54

