











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Pocket gophers (Rodentia: Geomyidae) and their chewing lice (Phthiraptera: Trichodectidae) represent a model system for the study of cophylogeny (Hafner et al. 2003, and references therein; Light and Hafner 2007). Geomyids are fossorial and asocial, occurring in patchily distributed populations, with genetically distinct groups being largely parapatric (Daly and Patton 1990; Hafner et al. 2003). Dozens of non-mammalian organisms inhabit the closed burrow system of pocket gophers and, like the pocket gophers, these organisms are also specialized in this unique niche. In fact, many of these parasites and inquilines are found nowhere else (Hubbel and Goff 1940; Tishechkin and Cline 2008). Among these pocket gopher-dependent organisms are over 100 named species and subspecies of chewing lice (Phthiraptera: Trichodectidae). The majority of these chewing lice seem to be highly host-specific, occurring on a single pocket gopher species or subspecies (Hellenthal and Price 1991). Chewing lice are wingless insects that feed on skin detritus of their hosts (Marshall 1981). The very specializations that make chewing lice well suited for a subterranean existence on a solitary host also greatly reduce their ability to disperse (Marshall 1981). When this poor dispersal ability is combined with the solitary nature of geomyids, the probability of colonizing a new host (host switching) is low (Hafner et al. 2003). Hence, chewing lice and their pocket gopher hosts are intimately associated. This intimate association continues across evolutionary timescales (Hafner et al. 1994; Light and Hafner 2007), making the pocket gopher-louse system a “textbook case” of cophylogeny (e. g., see Page and Holmes 1998; Ridley 2004; Futuyma 2005; Morris et al. 2016).
Although the Geomyidae have a distribution that stretches from Canada to northern Colombia (Hafner 2015), the family’s highest diversity (five genera) is found in the Trans-Mexican Volcanic Belt (TMVB). The TMVB consists of hundreds of volcanic structures stretching east to west within the Mexican Transition Zone between the Nearctic-Neotropical realms (Figure 1; Mastretta-Yanes et al. 2015). Aside from the striking physiographic relief of this 1,200 km belt, this region is characterized by a long history of volcanism beginning in the Miocene with major structural changes continuing through the Pleistocene and very recent formation of major stratovolcanoes (García-Palomo et al. 2000). In addition to this complex geological history, there have been at least three Pleistocene glacial-interglacial cycles (White 1986; Nixon 1989; Heine 1994a 1994b). These co-occurring events affected community composition and species phylogeography throughout the Pleistocene, and any genetic structure that came about during this period was likely the result of geology, climate, or more likely both (Mastretta-Yanes et al. 2015). Given the complex geologic and climatic history, one would expect a complex history of evolution in organisms distributed across this highly plastic landscape, and the genus Cratogeomys certainly meets this expectation. Prior to Hafner et al. (2004), six species of Cratogeomys were recognized from the area encompassed by the TMVB, distributed in a mosaic (Russell 1968). Cratogeomys merriami occupied territories from around Mexico City eastward to the east coast. Hafner et al. (2004) revised this to include three species comprising the C. merriami species group. Russell (1968) referred to Cratogeomys in the western half of the TMVB as the C. gymnurus species-group, and it was this species-group that most-reflected the complex history of the region with five named species and disjunct, mosaic distributions within C. tylorhinus and C. gymnurus. The complex geologic history, distribution of soils, and plasticity of the morphology in geomyids (Patton and Brylski 1987) undoubtedly made earlier morphological analyses problematic. Hafner et al. (2004) revised the C. gymnurus species group using to include two species, C. fumosus and C. planiceps comprising the C. fumosus species group (Figure 1). Cratogeomys planiceps is found in the vicinity of Nevado de Toluca Volcano and Valle de Bravo in central Mexico, and C. fumosus is broadly distributed across the western half of the TMVB and is composed of four genetically delineated clades that reflect the regions rich history (clades A-D; Figure 1).
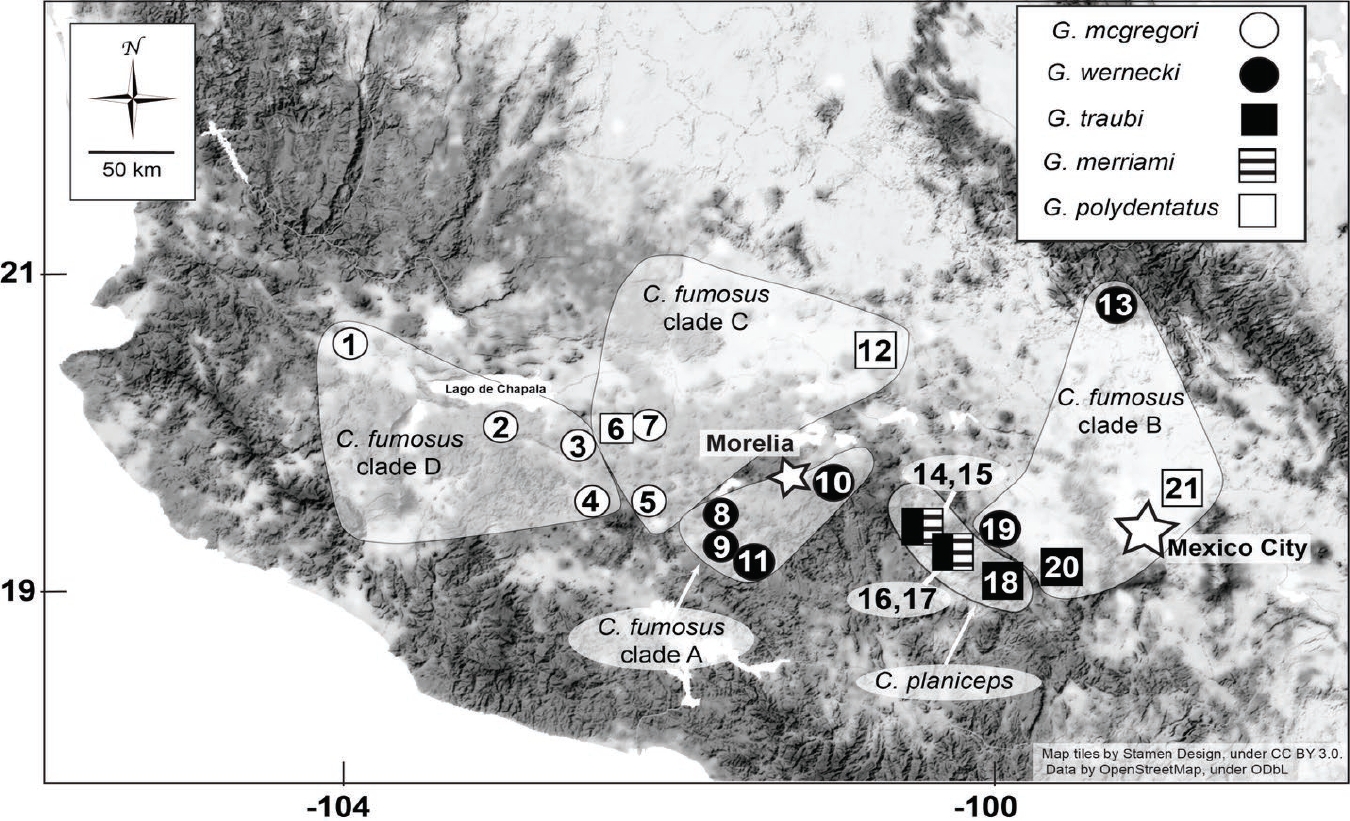
Figure 1 Map depicting distribution of the Cratogeomys fumosus species group and the five genetic clades delineated by Hafner et al. (2004). Symbols indicate localities and Geomydoecus louse species sampled for this study.
The known distribution of the chewing lice hosted by the C. fumosus species group is based on the definitive work of Price and Hellenthal (1989). The complex geologic and climatic history of the region along with likely contact with C. merriami has resulted in an interesting host-parasite assemblage involving three louse species complexes: the Geomydoecus mexicanus and G. coronadoi complexes (both also found on the C. merriami species group), and the G. mcgregori complex (shared with pocket gophers of the genus Pappogeomys). Of the five species of chewing lice included in this study, only G. traubi and G. merriami are also found on other taxa (both occur on C. merriami). Geomydoecus mcgregori is distributed across the western TMVB and is found on pocket gophers belonging to clades D and C (Price and Hellenthal 1989; Figure 1). Geomydoecus wernecki (a member of the G. mcgregori species complex) can be found on the remaining gophers of the C. fumosus species group (Figure 1). Geomydoecus polydentatus is a widely distributed taxon largely sympatric with chewing lice of the G. mcgregori species complex. One additional louse taxon, G. tellus was described from the northwestern extremes of the distribution of C. fumosus (e. g., locality 1, Figure 1; Price and Hellenthal 1988). This louse was morphologically distinct and has not been assigned membership to any existing species complex.
Paterson et al. (2003) discussed the kinds of historical events that will affect the degree of congruence between phylogenies of associated hosts and parasites. Whereas codivergence yields congruent phylogenies, four types of events can result in varying degrees of phylogenetic incongruence: 1) parasite duplication (speciation of parasite without speciation of host). 2) Parasite inertia (in which the host lineage diverges and the parasite lineage does not). 3) Host switching. 4) Lineage sorting. The asocial nature of pocket gophers and low vagility of chewing lice reduces the likelihood of host switching in this system, whereas lineage sorting is an intrinsic property of any host-parasite interaction. Gene trees that do not match species trees for the parasite lineage, the host lineage, or both can obscure any potential underlying pattern of cophylogeny. Moreover, an ancestral host population may carry multiple genetically divergent lineages of parasites, which can then be retained or lost in a stochastic manner on isolated host populations.
Demastes et al. (2012) demonstrated the effects that differing evolutionary scale can have on the presence of detectable cophylogeny in Pappogeomys and their chewing lice. Recently diverged lineages of hosts and parasites are less likely to show cophylogeny than older lineages because recently diverged populations of hosts are more likely to share gene flow (reticulate evolution of the host populations), which increases the likelihood of host switching and detracts from the likelihood of observing parallel phylogenies (Nieberding and Olivieri 2007). Successful transfer of parasites is more likely in hybridization events between intraspecific populations of hosts because the hosts are more similar (providing similar habitats for the parasites) and because there has been less time for the evolution of host specificity in the parasites. Incomplete lineage sorting also may obscure an underlying pattern of cophylogeny. This is a potential problem for phylogenetic analyses of recently diverged taxa, particularly when effective population size is large (Maddison and Knowles 2006). Because younger lineages have had less time for lineage sorting of parasites to occur, and because the genes we use to infer relationships in each lineage have had less time to coalesce, we are less likely to observe a pattern of similar evolutionary histories based on genetic analysis of more recently diverged pairs of host and parasite taxa (Hafner and Page 1995; Rannala and Michalakis 2003; Nieberding and Olivieri 2007).
Like Demastes et al. (2012), other studies of pocket gophers and chewing lice and of Neotropical figs and their pollinators have both shown less cospeciation at finer phylogenetic scales in lineages that appear to cospeciate on a larger scale (Demastes and Hafner 1993; Demastes et al. 2003; Jackson et al. 2008). These observations, however, are far from exhaustive. As Huyse et al. (2005) suggest, studies of multiple host-parasite systems at multiple taxonomic levels are needed before drawing any conclusions regarding the relationship between taxonomic level and likelihood of cospeciation.
Herein, we examine the chewing lice found on pocket gophers of the Cratogeomys fumosus species group using DNA sequence data from the mitochondrial cytochrome c oxidase subunit I (COI) gene. We use these data to compare the resulting genetic distance matrices and phylogenies for these lice with those of their hosts (Hafner et al. 2004) and test for the presence of a widespread pattern of cophylogeny.
Materials and Methods
Specimens examined. We examined a total of 21 specimens of Geomydoecus collected from the same specimens of pocket gophers (Cratogeomys), from 19 localities analyzed by Hafner et al. (2004). Specimens were collected under SEMARNAT Permit FAUT-0002 issued to F. A. Cervantes. Following DNA isolation, voucher specimens were preserved following Cruickshank et al. (2001) and identified to species based on the taxonomic characters of Price and Emerson (1971) and Price and Hellenthal (1989).
Phylogenetic Analysis. DNA extraction, amplification, and sequencing for chewing lice followed Light and Hafner (2007). Genomic DNA was extracted (DNeasy Tissue Kit, QIAGEN, Valencia, California) from individual chewing lice following the manufacturer’s protocol, with a final elution of 30 µl for each sample. Extractions were amplified by polymerase chain reaction (PCR) for overlapping regions of the mitochondrial cytochrome c oxidase subunit I (COI) gene, using primers LCO1490 and HCO2198 from Folmer et al. (1994) with an annealing temperature of 46 C for 40 cycles. Two additional primers were designed for this study, HCO758 (AAGAATATATACCTCTGGGTGACC) and HCO740 (GACCAAAAAACCAAAACAAATGC) to be used in conjunction with LCO1490. Success of PCR reactions was determined using agarose gel electrophoresis, and negative PCR controls were used to help detect any possible contamination that may have occurred pre-PCR. Resulting amplified fragments were prepared for sequencing using EXOSAP-it (USB, Cleveland, Ohio), and sequencing was performed at Iowa State University’s DNA Facility (Ames, Iowa) using their ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, California). All sequenced regions received at least 2x coverage by sequencing with both PCR primers. Sequences were aligned and edited using Geneious version 11.0 (Kearse et al. 2012). All sequences were subjected to a BLAST analysis using BLASTN version 2.8 (Zeng et al. 2000) to confirm target sequence. Sequences were submitted to GenBank (GenBank Accession Nos. KF005296 to KF005316). Geomydoecus bulleri (JF342595; from the G. bulleri species complex) was used as an outgroup to root all louse trees.
JModelTest version 2.1 (Guindon and Gascuel 2003; Darriba et al. 2012) was used to select nucleotide substitution models for maximum-likelihood and Bayesian analyses. Based on AIC (Akaike Information Criterion) criteria, the GTR + I + G model was used with the following model parameters: freqA = 0.40, freqC = 0.22, freqG = 0.13, freqT = 0.25, p-inv = 0.53, gamma shape parameter = 1.4. Maximum likelihood analysis was conducted using MEGA version 5.0 (Tamura et al. 2011). Support for nodes was estimated using 1,000 bootstrap replicates. MrBayes version 2.2 (Ronquist and Huelsenbeck 2003) was used for Bayesian analyses as implemented in Geneious version 11.0 (Kearse et al. 2012). Bayesian analysis consisted of paired runs of 4 Markov-Chain Monte Carlo analyses, each using default settings and iterated for 1.1 x 106 generations sampled every 200 generations, discarding the initial 110,000 trees (10% of total) sampled. Convergence was determined by examining the MCMC traces for proper mixing. Phylogenetic trees were visualized using FigTree version 1.4 (Rambaut 2012).
Tests of cophylogeny. The overall approach taken to test for cophylogeny followed the methods of Demastes et al. (2012) by using both matrix-based and topology-based analyses. Matrix-based tests for cophylogeny are topology-free and compare genetic distances for gene regions in the associated host and parasite taxa. Host genetic distances (uncorrected) were calculated from the 1,133 bp from the cytochrome b gene sequences from Hafner et al. (2004); Genbank AF302155, AF302156, AF302162, AF302163, AF302166-AF302170, AF302174, AF302175, AF302178-AF302183, AY545531-AY545533, AY545535, AY545539- AY545541) and compared to the uncorrected distances calculated for the chewing lice. Both distance matrices were calculated using the program MEGA 5.0 (Tamura et al. 2011). The null hypothesis of random association between the host and parasite distance matrices was tested using Mantel tests as implemented in the ape, version 5.0, package in R (R Core Team 2014; Paradis and Schliep 2018). Probability of a nonrandom association between the matrices was calculated using the standardized statistic (r) with 1,000 permutations. Tests were run on the complete 21 taxon dataset. Further tests were run with matrices that were pruned to only include chewing lice from the G. mcgregori species complex and their pocket gopher hosts. A second distance-based method, Parafit (Legendre et al. 2002), also was used to compare the distance matrices after converting them into principal component matrices. Tests of random association were conducted with 999 permutations globally and across all pairs of taxa. This analysis was performed using the Parafit option in the ape, version 5.0, package in R. Parafit tests were run on the complete 21 taxon dataset, and additional tests were run with matrices that were pruned to only include chewing lice from the G. mcgregori species complex and their pocket gopher hosts.
For topology-based cophylogenetic analyses, the host phylogeny was taken from Hafner et al. (2004) and pruned to include taxa hosting chewing lice from the Geomydoecus species complexes included in this study. The phylogenies of the hosts and parasites were compared to test for a history of widespread cophylogeny. Reconciliation analysis was performed using Jane version 4.0 (Conow et al. 2010) with a cost of zero for codivergence events and a cost of one for host switches, duplications, or losses. Significance was determined by randomization of the parasite tree (n = 10,000) and comparison of the resulting null distribution of codivergence events to number of codivergence events estimated from comparison of the data-based host and parasite trees. Analyses were performed on the full 21-taxa phylogeny and on subsets of the chewing louse species complexes.
Results
Because of the documented presence of at least two different louse species complexes at each of the sampled localities, there was the potential to obtain 38 distinct geographic samples (2 x 19 localities; Table 1). Despite examining multiple lice from each locality (and often multiple hosts individuals), we were only successful in identifying both possible lice from two localities, both within the C. planiceps distribution (Table 1). Therefore, at most sites, one of the two possible co-occurring species was absent from the host examined (Table 1).
Table 1 Specimens examined. Map column refers to Figure 1. “Geomydoecus complex” indicates which chewing louse species could be expected from each species complex on the Cratogeomys host listed compared with locality data from Price and Hellenthal (1989). Asterisks indicate the louse species actually identified and included in this study. Note that several additional lice from the same and from different hosts were examined from each locality but, to date, in no case have two species been found on the same host. LSUMZ = Louisiana State University, Museum of Natural Science.
| Geomydoecus complex | ||||||
|---|---|---|---|---|---|---|
| Map | Host | Collection # | Geographic Locality | mcgregori | mexicanus | coronodoi |
| 1 | C. fumosus | LSUMZ 36081 | Jalisco: 16 km E Ameca1 | mcgregori* | polydentatus | |
| 2 | C. fumosus | LSUMZ 36077 | Jalisco: 5 km SW Mazamitla | mcgregori* | polydentatus | |
| 3 | C. fumosus | LSUMZ 36164 | Michoacán: 5 km N Tingüindín | mcgregori* | polydentatus | |
| 4 | C. fumosus | CNMA 39673 | Michoacán: 2 km N, 5 km W Apo, 1,720 m | mcgregori* | polydentatus | |
| 5 | C. fumosus | LSUMZ 36133 | Michoacán: 2 km N Uruapan | mcgregori* | polydentatus | |
| 6 | C. fumosus | LSUMZ 36292 | Michoacán: 2 km NW Patamban | mcgregori | polydentatus* | |
| 7 | C. fumosus | LSUMZ 36124 | Michoacán: Tangancícuaro | mcgregori* | polydentatus | |
| 8 | C. fumosus | LSUMZ 34425 | Michoacán: 6.5 km S Pátzcuaro | wernecki* | polydentatus | |
| 9 | C. fumosus | LSUMZ 36136 | Michoacán: San Gregorio, 8 km E Opopeo | wernecki* | polydentatus | |
| 10 | C. fumosus | LSUMZ 36130 | Michoacán: 5 km S, 20 km E Morelia | wernecki* | polydentatus | |
| 11 | C. fumosus | LSUMZ 36129 | Michoacán: 1 km S Tacámbaro | wernecki* | polydentatus | |
| 12 | C. fumosus | LSUMZ 36092 | Guanajuato: 1 km E Celaya | wernecki | polydentatus* | |
| 13 | C. fumosus | LSUMZ 36091 | Querétaro: La Cañada, 9 km (by road) SW Pinal de Amoles | wernecki | polydentatus* | |
| 14 | C. planiceps | LSUMZ 36075 | México: 25 km N Valle de Bravo | traubi* | merriami | |
| 15 | C. planiceps | LSUMZ 36303 | México: 25 km N Valle de Bravo | traubi | merriami* | |
| 16 | C. planiceps | LSUMZ 36123 | México: 3 km S, 20 km E Valle de Bravo | traubi* | merriami | |
| 17 | C. planiceps | LSUMZ 36291 | México: 3 km S, 20 km E Valle de Bravo | traubi | merriami* | |
| 18 | C. planiceps | LSUMZ 34901 | México: 10 km S, 16 km W Toluca, 3,000 m | wernecki | traubi* | |
| 19 | C. fumosus | LSUMZ 36163 | México: 26 km W Toluca | wernecki* | traubi | |
| 20 | C. fumosus | LSUMZ 34902 | México: 1 km N La Isla, 2,612 m | wernecki | traubi* | |
| 21 | C. fumosus | LSUMZ 36066 | México: 1 km S Tepexpan | wernecki | polydentatus* | |
1 The morphologically unique Geomydoecus tellus has also been described from this locality and co-occurring on the same host individual as G. macgregori (Price and Hellenthal, 1988). It has not been assigned to a species complex and was not detected in our sampling.
Louse Phylogeny. For the 21 louse individuals included in this study, the 550 bp of COI sequence were generated and analyzed phylogenetically. The aligned dataset held 211 parsimony-informative and 329 invariable sites resulting in all 21 ingroup taxa possessing unique haplotypes. Maximum likelihood and Bayesian analyses produced phylogenetic trees with nearly identical topologies. Minor differences between maximum-likelihood and Bayesian trees for the lice disappeared when poorly supported nodes (less than 70 % bootstrap support or less than 0.8 posterior probability) were collapsed, producing a phylogeny that is largely concordant with current louse taxonomy (Figure 2). Genetic data revealed the presence of two genetically distinct lineages within G. mcgregori. Some of the louse clades recovered (Figure 2) are consistent with both geographic proximity and host taxonomy (Figure 1; G. mcgregori, localities 1, 2, and 4 and G. wernecki, localities 8, 9, and 11). However, one louse clade (G. mcgregori, localities 3, 5, and 7) includes geographic neighbors derived from hosts belonging to different genetic groups. Another louse clade (G. wernecki, localities 10 and 19) includes specimens that are not in geographic proximity and that occur on hosts belonging to different genetic groups. Likewise, other closely related lice (for example, G. polydentatus, localities 12 and 21 and G. traubi localities 16 and 20) are found on geographically distant hosts belonging to different genetic groups.
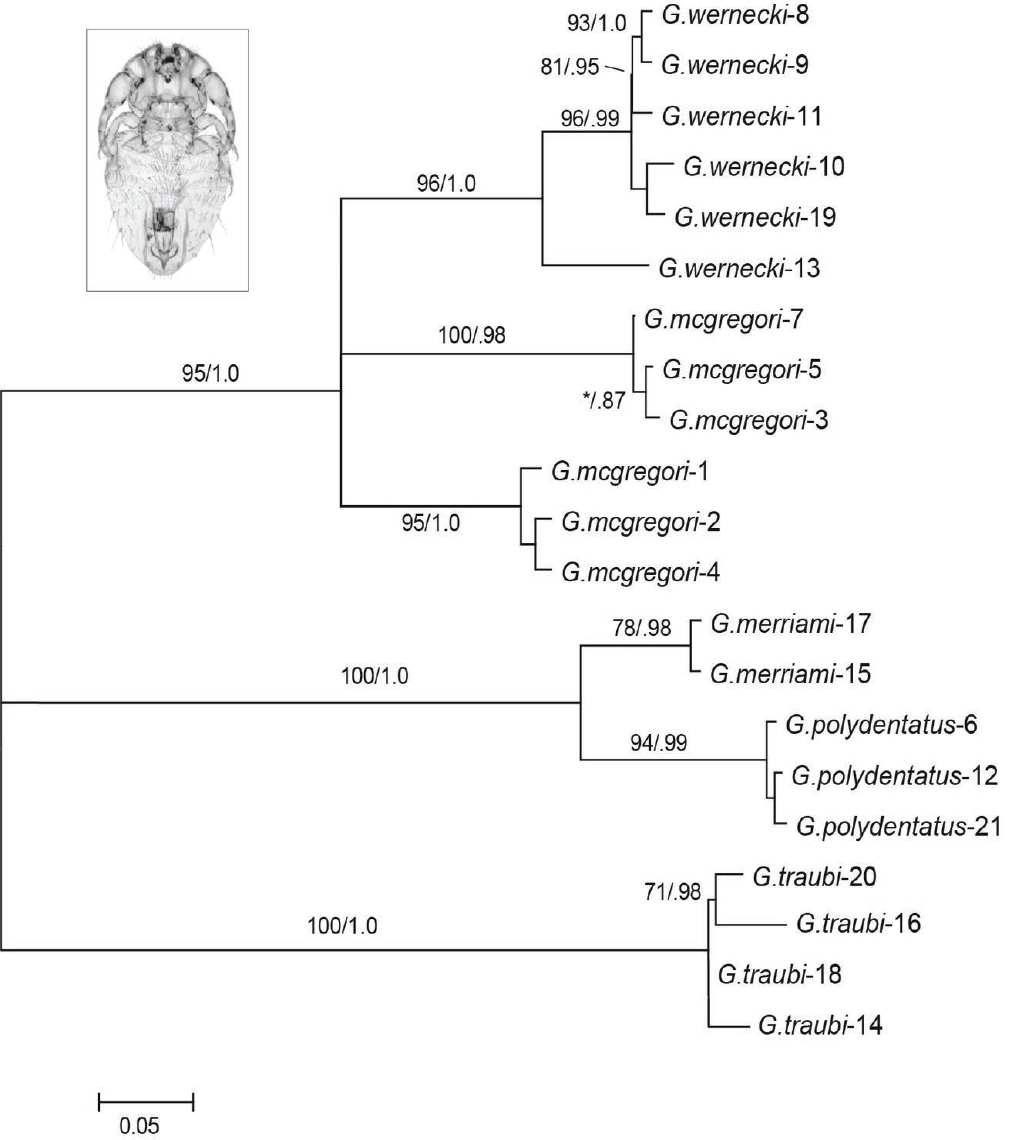
Figure 2 Maximum likelihood (ML) tree for 21 Geomydoecus lice studied (G. mcgregori in inset). Locality numbers refer to Figure 1 and Table 1. Numbers on branches indicate ML bootstrap support/Bayesian posterior probability.
Tests of Cophylogeny. Matrix comparison for genetic distances of chewing lice and pocket gophers indicated a significant correlation for both the complete dataset (Mantel test, z = 3.19, P = 0.007) and for matrices trimmed to include only the clades involving the G. mcgregori complex (z = 0.68, P = 0.005). Similar results were returned using the Parafit algorithm. Global values for the complete dataset (F = 0.006, P = 0.008, 10 of 21 links significant) and the trimmed version (F = 0.007, P = 0.004, 7 of 12 links significant) also indicated a significant positive correlation for the host and parasite distance matrices.
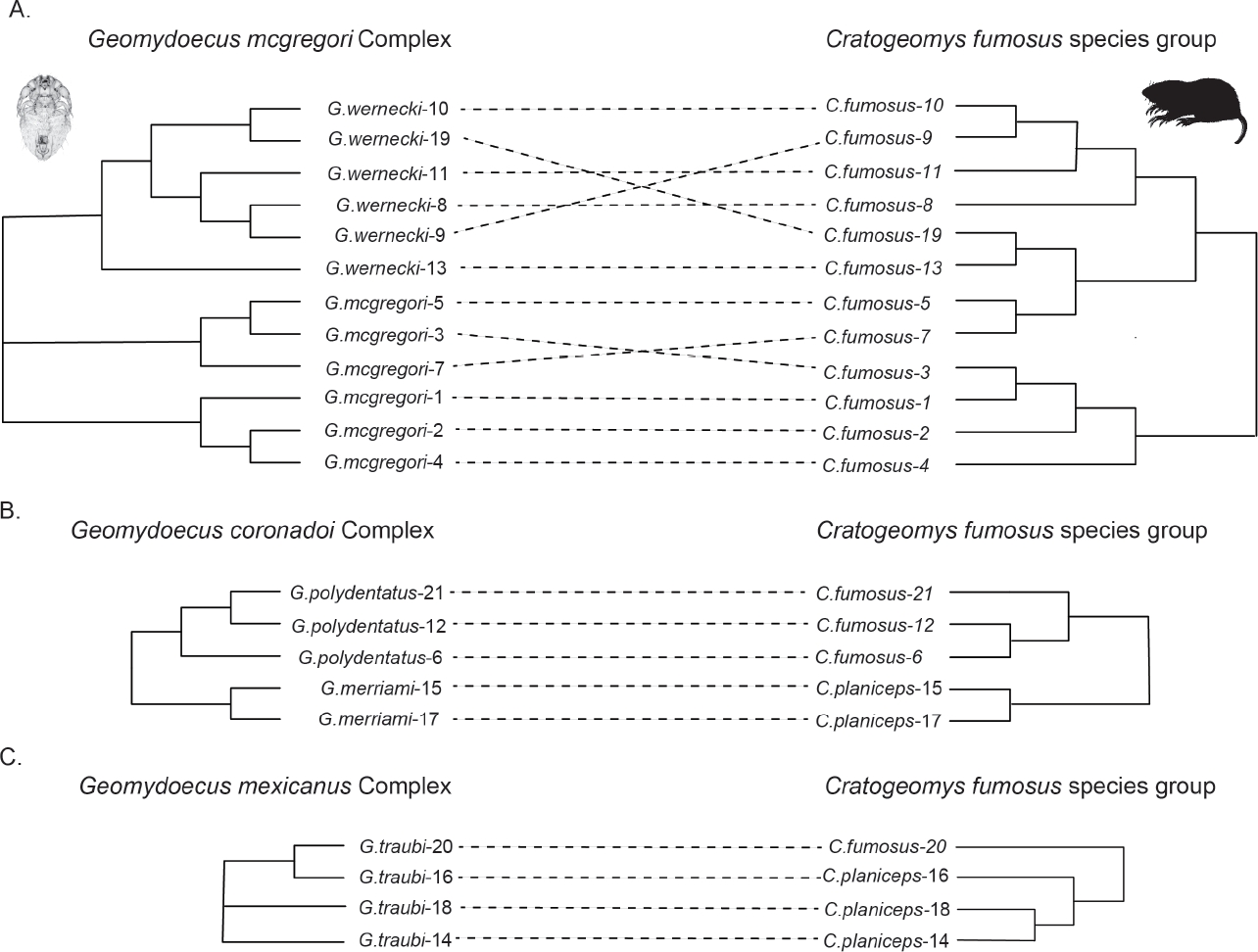
Tanglegram comparison superficially indicated little similarity between host and parasite phylogenies (Figure 3). Cophylogenetic reconstruction analysis using Jane (Conow et al. 2010) indicated the absence of a widespread pattern of cophylogeny for the chewing lice and pocket gophers. In that analysis, twenty-eight percent of the cophylogenetic reconstructions using the Hafner et al. (2004) gopher topology and a randomly generated topology for the chewing lice had costs that were equal to or less than the cost of the best (lowest cost) reconstruction for the observed data (cost of observed topologies = 16; P = 0.3). Analyses were repeated for each chewing louse species complex (Figure 4). These analyses for the G. mcgregori, G. coronadoi, and G. mexicanus complexes also indicated the lack of a widespread nonrandom pattern of cophylogeny (P = 0.3, P = 0.5, and P = 0.7 respectively). However, some of these comparisons were hindered by small sample sizes and, short of perfect congruence, were not likely to indicate statistical significance.
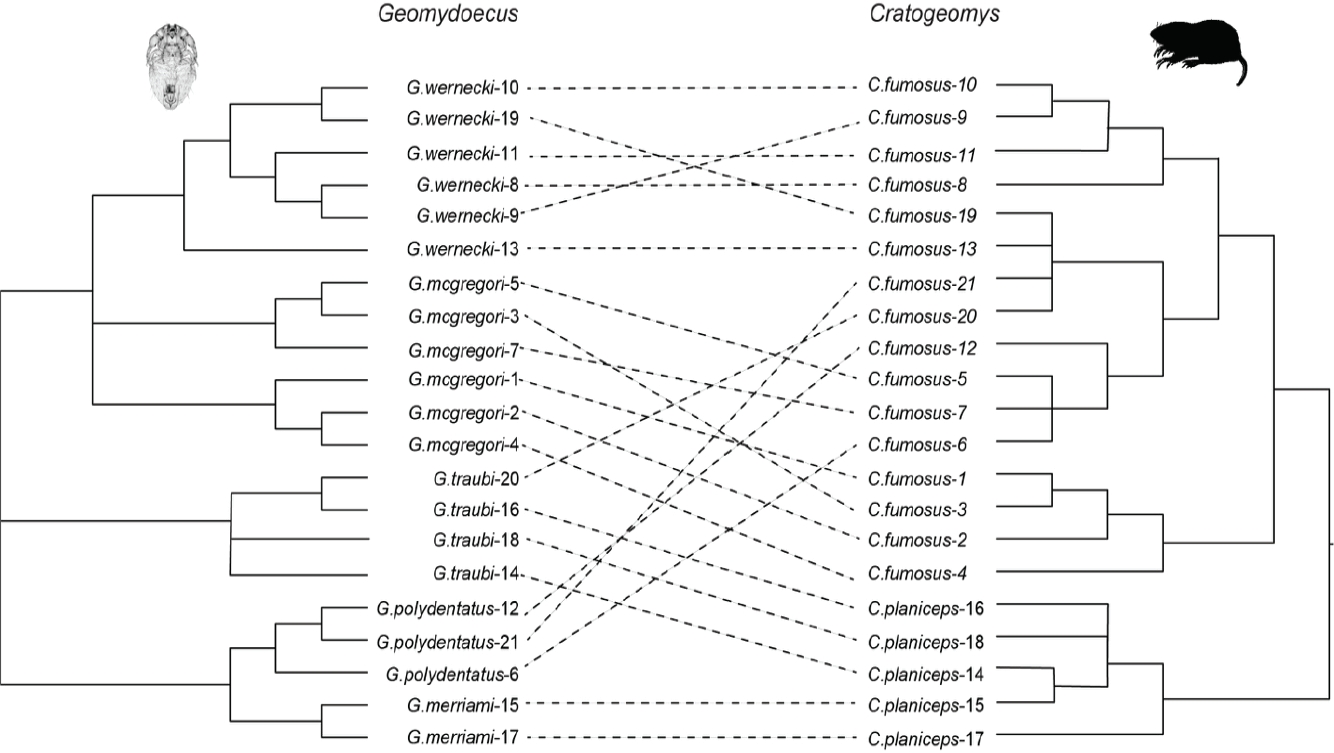
Figure 3 Tanglegram illustrating host parasite associations between pocket gophers (Cratogeomys) and their chewing lice (Geomydoecus). The pocket gopher phylogeny is taken from Hafner et al. (2004) and pruned to include only pertinent specimens.
Discussion
Louse Phylogeny. Past genetic examinations of morphologically-based phylogenies of chewing lice of pocket gophers have revealed a remarkable level of accuracy in the earlier taxonomic studies, even at the subspecies level (e. g., Hafner and Nadler 1988; Demastes and Hafner 1993; Light and Hafner 2007). Our molecular data indicated genetically cohesive groups of individuals consistent with the morphologically-based species-level taxonomy of Price and Hellenthal (1989), with the exception that they reveal for the first time the existence of two divergent, well-supported and potentially paraphyletic clades of lice within G. mcgregori. Although these two G. mcgregori clades (localities 1, 2, 4 and localities 3, 5, 7) are not concordant with their associated host phylogenies, they do occur as geographically cohesive units (Figure 1).
Tests of Cophylogeny. As reviewed by de Vienne et al. (2013), there are distinct advantages and disadvantages to both matrix-based and reconciliation-based methods for the testing of cophylogeny. One drawback for matrix-based methods such as Mantel tests and Parafit is their inability to account for phylogenetic non-independence (Felsenstein 1985). This in itself could account for a possible false-positive signal of cospeciation from matrix-based approaches. While matrix-based approaches indicated significant congruence between louse and host genetic distances in this study, the more rigorous reconciliation analyses suggested no significant pattern of cophylogeny. Relatively few nodes are shared between host and parasite trees, indicating a large degree of discordance between host and parasite phylogenies, with several closely related louse specimens occurring on rather distantly related hosts (Figure 3).
Past studies have established that pocket gophers and chewing lice frequently show a pattern of cospeciation, an outcome likely favored by the life histories of both lineages (e. g., Hafner et al. 2003; Light and Hafner 2007; Demastes et al. 2012). The pattern of non-cospeciation observed here may be the result of several factors. First, results in this study come from a portion of a single mitochondrial gene. While the pivotal nodes of the parasite tree are well supported in our analyses, it is possible that a more robust analysis with additional genetic loci from the nucleus could reveal a different pattern of relationships that reflect an underlying pattern of cospeciation if incomplete lineage sorting in the mitochondrial data has incorrectly reflected the true phylogenetic history of the group. Secondly, several potential pairs of co-occurring chewing lice were not sampled, either because they are rare or because they have been extirpated from the sampled localities. Loss of these populations could obscure a pattern of cophylogeny if extirpation was followed by colonization by a new form. The G. wernecki at locality 3 (Tingüindín, Michoacán) may be the result of such an extirpation/colonization event given that the phylogenetic position of this louse (Figure 3) is more consistent with geography than with host relationships (Figure 1). It is also possible that the recent age of this particular system may play a role in the lack of cospeciation observed here by facilitating the likelihood of successful host switching and/or through the influence of lineage-sorting effects (Hafner and Page 1995; Rannala and Michalakis 2003; Nieberding and Olivieri 2007; Demastes et al. 2012).
Finally, as discussed previously (and in detail in Demastes et al. 2002; pp. 150-152) the Quaternary history of the western TMVB is quite complex with major geologic events occurring after estimated times of speciation for the hosts included in this study, C. fumosus and C. planiceps. The distributions for the five differentiated clades of pocket gophers included here (Hafner et al. 2004) likely experienced major changes during periods of increased volcanism and climatic shifts in the Pleistocene and Holocene. This complex history of shifting distributions caused by natural events would have presented many opportunities for contact between host individuals of different genetic groups and subsequent host switching of parasites (Paterson et al. 2003). The complex, dynamic history of the western TMVB has been the driving force behind the generation of the unique diversity of the region (González-Fernández et al. 2018; Mastretta-Yanes et al. 2015; Navarro-Sigüenza et al. 2007). As more phylogeographic studies are conducted on co-distributed taxa of the TMVB, a comparative approach may provide greater insights into the effects these events had on the biota of the region. For example, an alligator lizard (genus Barisia) exhibits a pattern of geographic differentiation that is very similar to that of Cratogeomys across the TMVB (Bryson and Riddle 2012). It is possible that frequent range changes resulting from climate change in this geologically dynamic region have generated an assemblage of pocket gophers and chewing lice with frequent opportunities for host-switching. This, coupled with a great potential for isolation and local extinction of chewing lice in the geographically intricate TMVB, could have caused obscured the normal pattern of cophylogeny typically observed for geomyids and their trichodectid lice.

