



![In silico, Synthesis and Biological Investigations of Pyrrolo[3,4-C]Pyrrole Hydroxamic Acid Derivatives as Potential Anticancer Agents](/img/es/prev.gif)







 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introduction
In previous twenty years, many synthetic efforts have been done to design organic semi-conductors for organic solar cells(1, 2). Major advantage of organic solar cells is cheap and easy device fabrication methods.
Phenyl-C61-butyric acid methyl ester(PCBM) is still unbeatable acceptor with high power conversion efficiencies resulted from due to its strong tendency to accept electrons from donor semiconducting polymers3. At the same time, poor absorption in visible region of solar spectrum PCBM and other fullerene derivatives stimulate the scientists to develop inexpensive alternate acceptors having qualities of PCBM as well as remove disadvantages of PCBM.
Organic acceptors can be divided into two sub-groups, polymers and small molecules. Polymer acceptors with respect to PCBM, show good absorption in the visible region and high open circuit voltages due to higher lowest unoccupied molecular orbital (LUMO) than PCBM. They also have disadvantages, give polydispersedmaterials due to irregular molecular weight from batch to batch, resulted low organic photovoltaic (OPV)performance4. Also show smaller fill factors due to limited electron transport caused by various deep traps and defects in acceptor polymer networks5. It is need in deed to explore more alternative small molecule acceptors (monodispersed).
Mostly scientists tried to design the acceptors with electronic and optical properties compliment with poly(3-hexylthiophene) (P3HT) donor6, which can be easily synthesized on large scale. To get good performance donor and acceptor should have complementary electronic and optical properties. Thus it is important to understand that how the optical as well as electronic properties can be tuned by structural tailoring to get significant improvement.
Absence of suitable matching between absorption spectrum of the active layer and the solar spectrum is one of major limiting factors of organic solar cells. Recently, near-IR small molecules base organic solar cells (OSCs) have gained increasing attention as indicated by published review articles7,8,9,10,11. For the better utilization of solar spectrum and to get improved efficiency, it is critical requisite to design and synthesize low bandgap small molecules with intense absorption in visible and near-IR regions12.
Research and development of new electron acceptors with lower LUMO levels than the commonly used PCBM is also needed for achieving efficient solar cells. In recent years, many investigations on the design and synthesis of low-band gap electrochromic polymers have been the major attractive area and a most successful and flexible strategy to design small band gap polymers involves the alternation of electron-rich and electron deficient units along the polymer chain 13,14,15,16. Some of this type polymers, quinoxaline units were used as acceptor unit and some works reported that low-bandgap polymers containing quinoxaline units as acceptor unit performed good electrochromic ability 17,18,19,20,21,22,22,23,24,25,26. These polymers generally show a variety of excellent properties such as distinct optical changes throughout the visible region, fast switching time, low-voltage electrochromic (the voltage lower than 1.3 V). To achieve high PCE values an ideal bandgap of 1.2-1.6 eV is necessary 27.
Thanks to computational methods, that it is possible to study large numbers of compounds even before synthesis. So quantum chemical methods can be used to study the series of compounds to establish the structure-efficiency relationship. Keep this in mind;we have designed a series of compounds with Acceptor-Conjugated system-Central block-Conjugated system-Acceptor. During this study, we have used one central building block. Different conjugated systems and terminal acceptors were used.
In this study, a series of small molecule acceptors were designed. Different π-spacer and terminal acceptors were used to design low band gap acceptors. Quantum chemical methods were used to predict the electronic and optical properties of designed compounds.
2. Computational Details
Previously, it has been showed that B3LYP functional provide the good results among standard DFT28,29,30,31,32,33,34 functionals for geometry optimizations of medium and large size molecules 35,36,37,38,39. The B3LYP functional and 6-31G(d,p) basis set was successfully used to optimize the ground state geometries where it was found a suitable level for small organic compounds40, 41. Moreover, Huong and coworkers stated that the B3LYP/6-31G(d,p) level is decent to imitate the experimental geometries42. The B3LYP wasextensivelyusedfor organic compounds to calculate the structural, electronic, charge transfer and optical properties which reproduces the experimental data significantly 33, 43,44,45,46. The 6-31G(d,p) basis set is balance for accuracy and computational time. Success of this level of theory is already proven for organic solar cell molecules. Geometries of all compounds in ground state were optimized using density functional theory (DFT) at B3LYP/6-31G(d,p) level. In addition, all geometries examined in this work were subsequently examinedthrough harmonic vibrational frequency calculations to verifytheir position on the B3LYP/6-31G(d,p) potential energy surface. A proper theoretical method isneeded to calculate the UV/vis spectrum of molecules, selection of suitable method become more important for compounds with high charge transfer (CT) character. To simulate the UV/vis spectra, use of time-dependent DFT (TD-DFT) is good choice.
Arago et al.,47, 48 have performed the benchmark studies using wB97X-D functional against several dispersion-corrected hybrids and highly correlated methods. They found that the wB97X-D exhibits the best performance to deal with charge-transfer excitations in a balanced way. The wB97X-D functional uses a version of Grimme’s D2 dispersion model. Thus we have carried out benchmark studies on three acceptors (one from each phase), i.e., CS1TA4, CS2TA4 and CS3TA4 to investigate the absorption wavelengths using wB97X-D functional. Furthermore, the methanol as solvent was used to estimate the absorption wavelengths. The polarized continuum model (PCM) was used to calculate the bulk solvent effects for all stages 49, 50. The calculated absorption wavelengths at TD-CAM-B3LYP/6-31(d,p) level in gas phase, TD-CAM-B3LYP/6-31(d,p) level in Methanol and TD-wB97X-D/6-31(d,p) level in Methanol have been tabulated in Table 1. By comparing the absorption wavelengths at TD-CAM-B3LYP in gas phase and solvent phase, it can be found that there is no significant effect of solvent, i.e., 6, 24 and 9 nm differences for CS1TA4, CS2TA4 and CS3TA4 between both the phases (gas and solvent at TD-CAM-B3LYP), respectively. Additionally, we have compared the absorption wavelengths computed at TD-CAM-B3LYP/6-31(d,p) level in gas phase and TD-wB97X-D/6-31(d,p) level in solvent. Interestingly, the careful analysis of Table 1 showed that the absorption wavelengths computed at TD-CAM-B3LYP/6-31(d,p) level in gas phase and TD-wB97X-D/6-31(d,p) level in Methanol have no significant effect, i.e., the difference in the absorption wavelengths is 12, 10 and 9 nm for CS1TA4, CS2TA4 and CS3TA4, respectively. Thus from this benchmark study, it can be expected that the absorption wavelengths computed at TD-CAM-B3LYP/6-31(d,p) level in gas phase are comparable to the TD-wB97X-D/6-31(d,p) level in Methanol. So in the present study all the TD calculations have been performed by using TD-CAM-B3LYP/6-31(d,p) level in gas phase.
3. Results and discussion
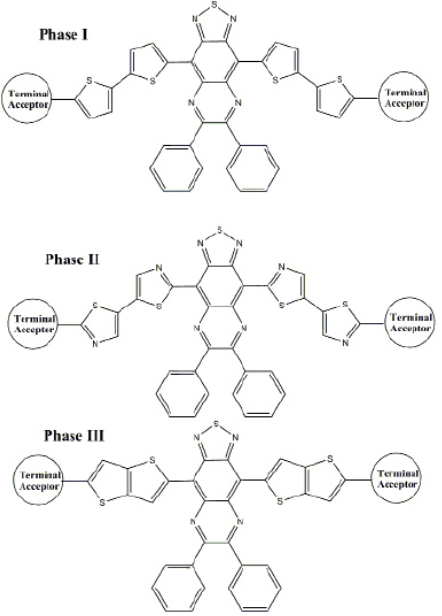
In the course of this study, a series of acceptors weredesigned using different π-spacer and terminal acceptors (Fig. 1). Designing framework was based on three phases,in every phase different π-spacer with all acceptors (TA1-TA5) wereused (Fig. 2). Only one core-building block wasused (Thiadiazolo3,4-gquinoxaline). This block was used with the aim to design low band gap molecular acceptors.

3.1. HOMO energy levels and band gaps
Solar cell efficiencies are closely related to frontier molecular orbital (FMO) energies and band gaps (Eg) 27. The VOC and driving force for exciton dissociation can be predicted. The energy difference between the LUMO of the electron acceptor and the HOMO of the electron donor was found to closely correlate with the Voc value 22,24,25. While driving force for exciton dissociation electron is equal to energy difference between LUMOs of the donor and the acceptor. In this study DFT was used to calculate the energy levels.
To study the structural effect of terminal acceptorgroups, we have used same conjugated system and core-building block. Thiadiazolo3,4-gquinoxaline as a core building block and bithiophene as the conjugated bridgewasused. This strategy allow to compare the effect of terminal acceptors51.
The results of phase I theoretical experiments provided insight for the selection of the most appropriate terminal acceptor based on the aforementioned criteria. For phase I HOMO and LUMO energies range is from -5.51 to -5.13 eV and from -4.22 to -3.90 eV, respectively. From Fig. 3, it is clear that acceptor groups have significant effect on energy levels. So modification of terminal acceptor is good way to make suitable energy level match between donor and acceptor that is necessary to achieve high efficiency. The CS1-TA2, CS1-TA4 and CS1-TA5 show similar energy levels. All the acceptors have nearly similar energy gap because core building block is same. As we all know, narrowing HOMO-LUMO gap of a molecule can harvest more sunlight, which leads a higher JSC 10. All terminal acceptors have same effect on HOMOs and LUMOs energy level. Indan-1,3-dione (TA2) based Indacenodithiophene small molecule showed efficiency higher than 6% against 6T donor. Results of this study showed that N-phthalimide (A3) terminal acceptor displays high lying LUMO than TA2. ByusingTA3 as terminal acceptor in above mentioned compounds, we can practice these compounds against easy synthesizable donor P3HT52. This idea supported by the work of Hou et al. They fabricated small molecule acceptor ITIC (i.e., TA2) with P3HT and PDCBT. It is very surprising to see that although these two polymers are all ideal donor materials for the PSCs using PC71BM as acceptor, the blends of P3HT:ITIC and PDCBT:ITIC exhibited very different photovoltaic properties. The PDCBT:ITIC based solar cell showed a efficiency of more than 10%, while surprisingly, P3HT:ITIC showed very less efficiency (1.25%)53. Low efficiency can be explained on the basis of high lying LUMO of P3HT. So, it is necessary to modify the acceptor using different terminal groups to suitable energy map with easily synthesizable and cheap donor (P3HT). The TA2 and TA5 showed almost similar energy levels. Replacement of TA2 with TA5 in above already reported compounds can give similar performance.
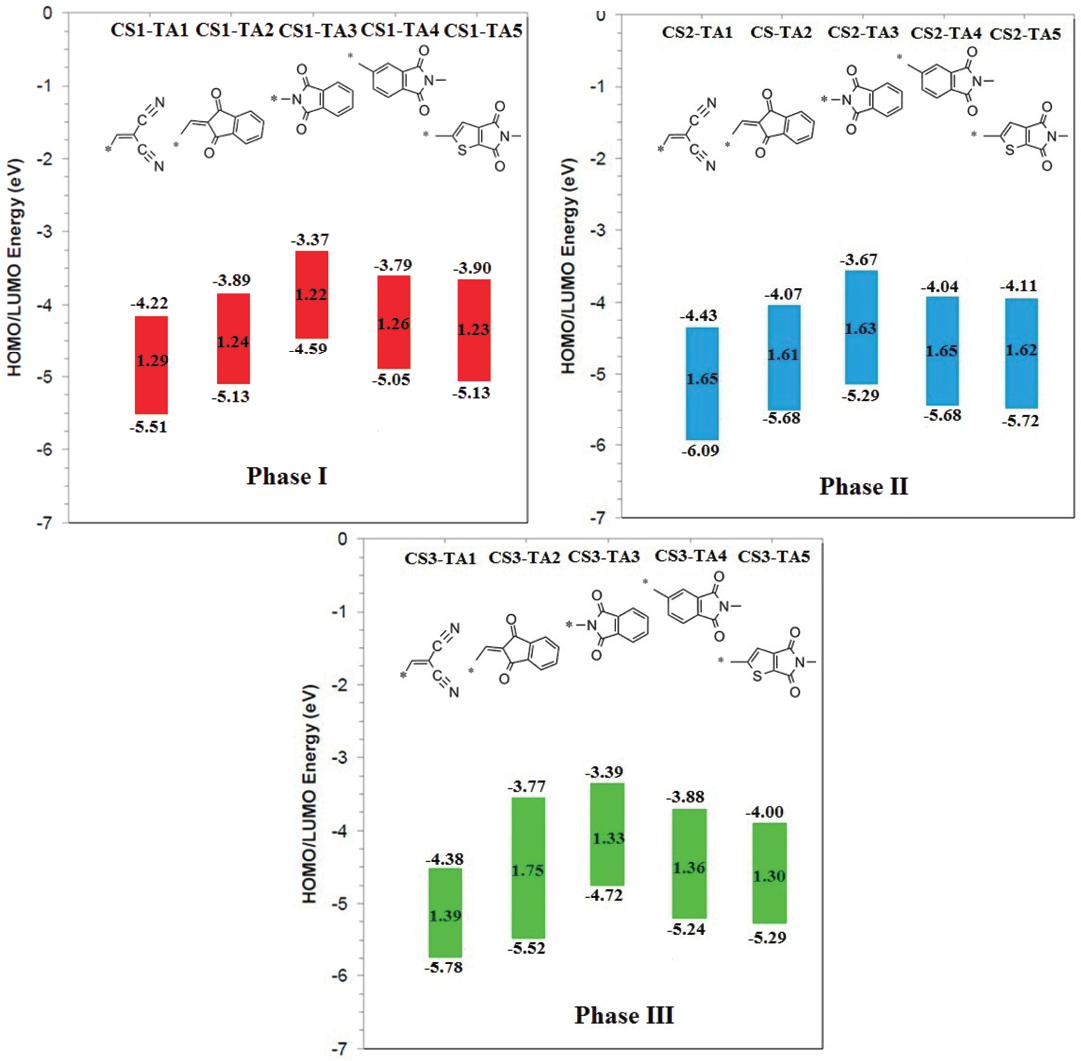
For phase II, thiazole based conjugated bridge was used. The increase in the energy gap wasobserved but difference was not so significant. The HOMO and LUMO energy level trend was also same. For Phase III, thieno3,2-bthiophene was used as conjugated system. The energy gap wasless as in Phase I, only CS3-TA2 showed large energy gap. Using different π-spacers, we can modify the HOMO and LUMO energy levels to get suitable spectral response.
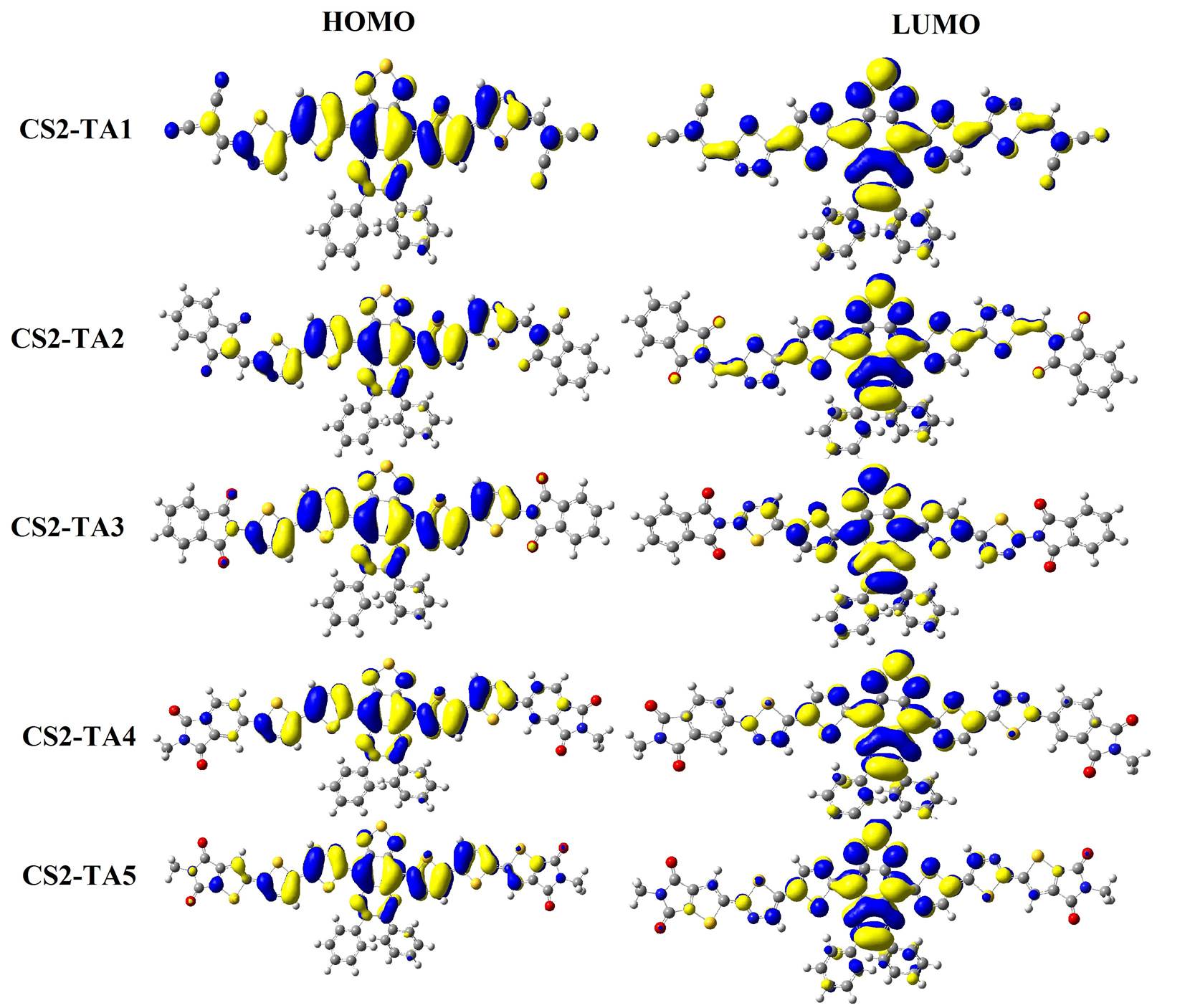
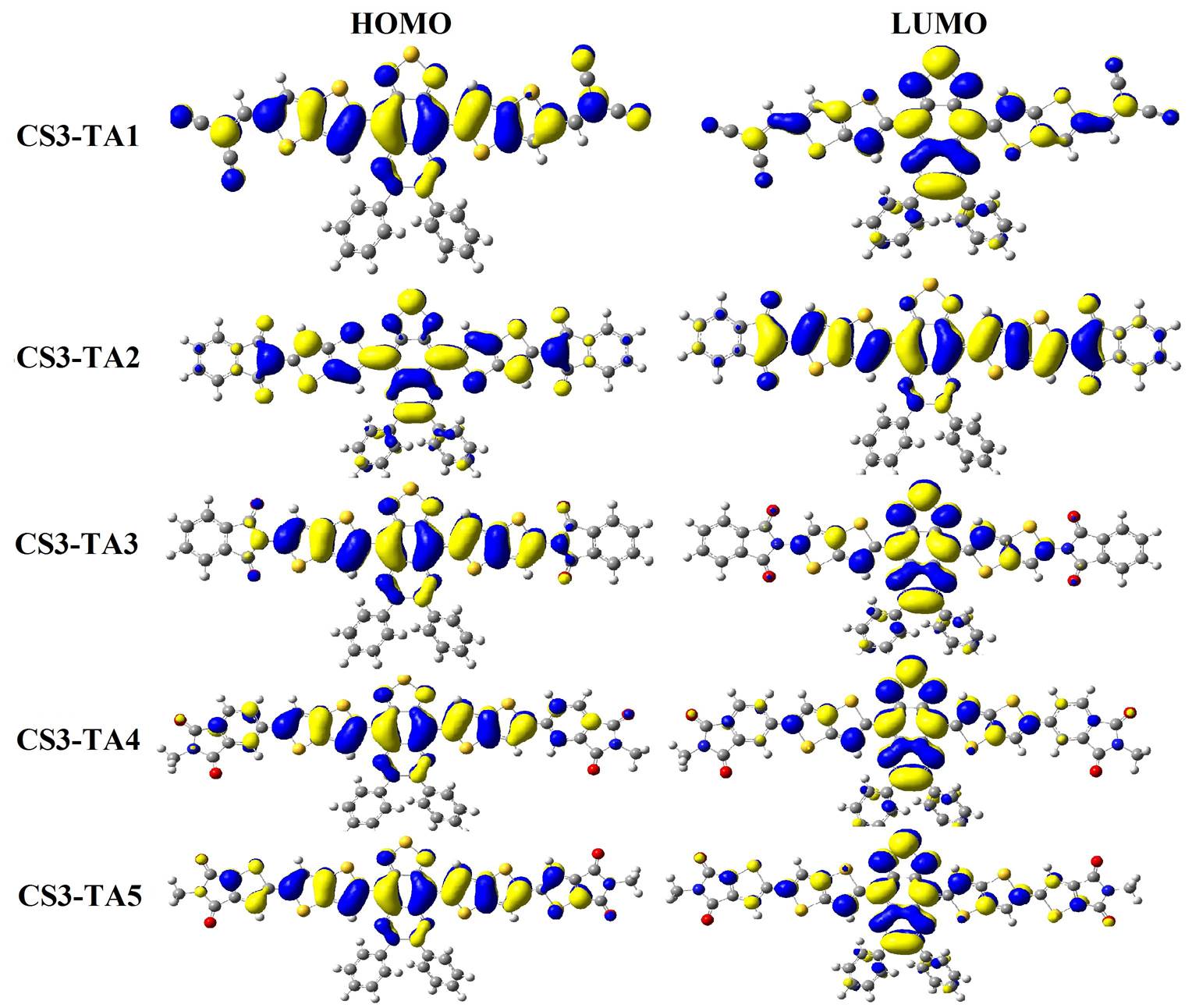
Graphical presentation of HOMOs and LUMOs is given in Figures 4, 5 & 6 for acceptors designed in three phases. The HOMOs and LUMOs charge density was found on main backbone which indicates that all the molecules are good acceptors.

3.2. Absorption Spectra
Absorption behavior of acceptor is also important like donor. It influences the efficiency of solar cell. Broad and intense absorption spectrum that matches with spectrum of donor is required. Absorption spectra of acceptors designed in three phases are given in Figures 7, 8 & 9. Absorption parameters are given in Table 2. For phase I, change of terminal acceptor showed reasonable change in absorption energy and absorption wavelength. The CS1-TA3 showed lower absorption wavelength. For phase II, all the acceptor absorb in higher wavelength. Modification of π-spacer is good to modify the absorption profile. For Phase III absorption wavelength was found blue shifted. But as compare to phase I, their absorption wavelength is red shift, especially CS3-TA3 and CS3-TA5 show higher wavelength (861 and 854 nm, respectively). It is generally accepted that acceptor show highextinction coefficient as compare to fullerene-based acceptors. In phase I, CS1-TA2 and CS1-TA4 showed higher oscillator strength while in phase II, CS2-TA1 and CS2-TA2 showed higher values. For phase III, results were found in similar trend. But CS3-TA2 show remarkably higher oscillator strength (3.32). This extends the conjugation of the aromatic system, reducing the bandgap significantly to give an absorption profile that is more complementary to that of P3HT.
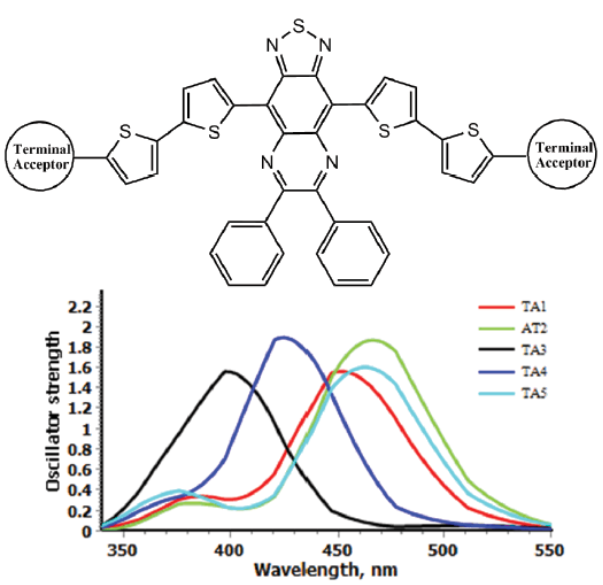
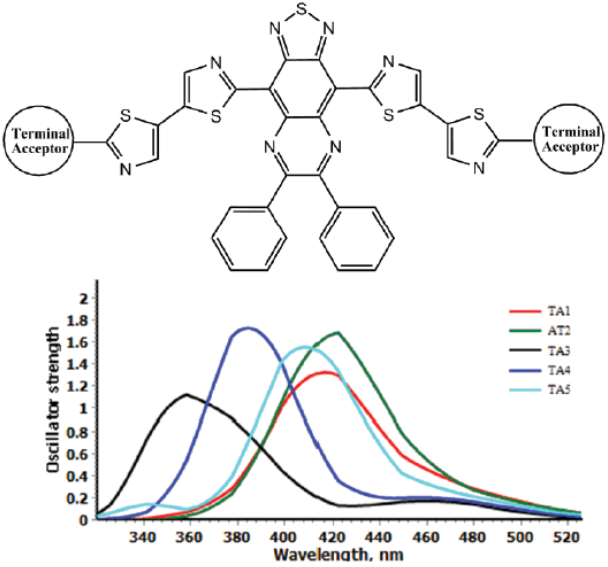
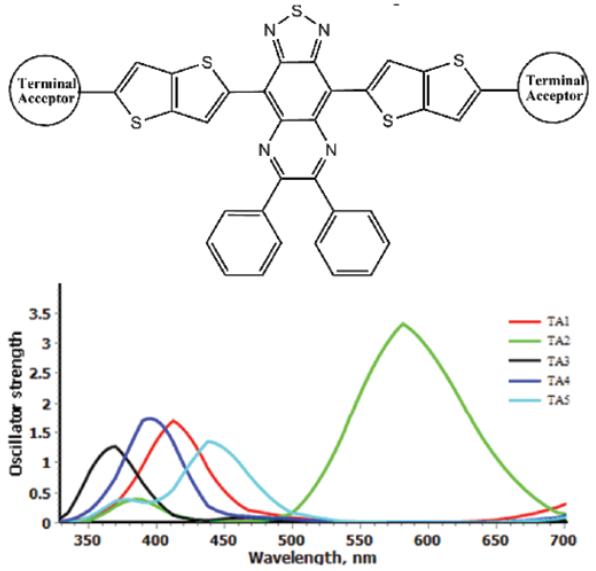
Table 2 Computed maximum absorption wavelengths (λmax/nm), absorption energy (Eg / eV), oscillator strengths (f), light harvesting efficiency (LHE)and transition natures of acceptors at TD-CAM-B3LYP/6-31(d,p) level of theory.
| Acceptor | E (eV) | λ (nm) | f | LHE | electronic configurations | |
|---|---|---|---|---|---|---|
| Phase I | CS1-TA1 | 2.72 | 455 | 1.37 | 0.957 | HOMO->L+2 (64%) |
| CS1-TA2 | 2.67 | 465 | 1.92 | 0.988 | HOMO->L+2 (72%) | |
| CS1-TA3 | 3.07 | 404 | 1.45 | 0.965 | HOMO->L+3 (47%) | |
| CS1-TA4 | 2.90 | 427 | 1.89 | 0.987 | HOMO->L+2 (47%) | |
| CS1-TA5 | 2.68 | 462 | 1.67 | 0.979 | HOMO->L+2 (57%) | |
| Phase II | CS2-TA1 | 1.81 | 687 | 1.20 | 0.937 | HOMO->LUMO (94%) |
| CS2-TA2 | 1.76 | 705 | 1.33 | 0.953 | HOMO->LUMO (94%) | |
| CS2-TA3 | 1.79 | 693 | 0.89 | 0.871 | HOMO->LUMO (96%) | |
| CS2-TA4 | 1.80 | 688 | 1.06 | 0.913 | HOMO->LUMO (95%) | |
| CS2-TA5 | 1.79 | 694 | 1.18 | 0.934 | HOMO->LUMO (94%) | |
| Phase III | CS3-TA1 | 2.98 | 416 | 1.40 | 0.960 | HOMO->L+2 (74%) |
| CS3-TA2 | 2.13 | 383 | 0.40 | 0.602 | HOMO->LUMO (92%) | |
| CS3-TA3 | 1.44 | 359 | 0.75 | 0.822 | HOMO->LUMO (98%) | |
| CS3-TA4 | 1.44 | 397 | 1.02 | 0.905 | H-2->LUMO (31%) | |
| CS3-TA5 | 1.45 | 854 | 1.01 | 0.902 | HOMO->LUMO (97%) |
The light absorption range plays a very important role on the Jsc. The Jsc can be expressed as54:
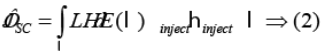
In above equation LHE is harvesting ability, Фinjectis the overall charge collection efficiency, and ηcollect. the overall electron injection efficiency. LHE can be calculated using following equation 55.
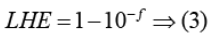
Tuning the absorption spectra is very important to get high Jscvalues. Generally, it is accepted that to get high conversion efficiency, high Jsc values is required. By modifying the π-spacer and terminal acceptor, we can alter the spectral profile.
Conclusion
In seekingto design narrow-band-gap small molecule acceptors, theoretical investigation was carried out. Most of the small molecules showedsmaller band gap. The frontier molecular orbital levels indicate that we can use large number of donors. It was also found thatπ-spacer modified the energy gap and UV/vis profile. Terminal acceptor TA2 showed efficiency up 11% that is reported. The TA2 and TA4 have almost similar energy level and behavior; these can be used to design efficient photovoltaic materials.

