










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.59 no.4 Ciudad de México oct./dic. 2015
Article
Quinone Redox-active Ionic Liquids
Andrew Patrick Doherty,*1 Sean Patterson,1 Laura Diaconu,1 Louise Graham,1 Rachid Barhdadi,2 Valentin Puchelle,2 Klaudia Wagner,3 David L Office3 and Jun Chen3 and Gordon G Wallace3
1 School of Chemistry and Chemical Engineering, The Queens's University of Belfast, Belfast, Northern Ireland, BT9 5AG, United Kingdom. e-mail a.p.doherty@qub.ac.uk, telephone + 44 (0)2890974481, fax + 44 (0)28906524.
2 Université Paris-Est Créteil, IEES Paris - DIIM, 61, Avenue du General de Gaulle, 94010 CRETEIL Cedex, France.
3 Intelligent Polymer Research Unit, ARC Centre of Excellence for Electromaterials Science, University of Wollongong, Wollongong, NSW, Australia.
Received November 17th, 2015;
Accepted January 18th, 2016.
Abstract
Simple ionic liquids exhibit unique physical and chemical properties that make them very useful for deployment in electrochemical devices such as solvent-free electrolytes in capacitors and batteries. However, incorporating redox functionality into ionic liquid structures opens up in situ faradaic electrochemistry which allows access to a large array of new electrochemical applications reliant upon heterogeneous or homogenous electron-transfer processes. This paper presents and discusses the opportunities and challenges for these types of electro-materials across a myriad of applications by considering exemplar quinone-functionalised ionic liquids.
Key words: task specific ionic liquids; redox active; applications; electrochemical devices.
Resumen
Los líquidos iónicos simples exhiben propiedades físicas y químicas únicas, lo que los hace de utilidad en dispositivos electroquímicos, funcionando como electrolitos libres de disolvente en capacitores y baterías. Sin embargo, la incorporación de funcionalidades redox en las estructuras de líquidos iónicos permite realizar electroquímica faradaica in situ, abriendo una nueva categoría de aplicaciones electroquímicas relevantes como procesos de transferencia electrónica heterogénea u homogénea. Este trabajo presenta y discute las oportunidades y retos en la generación de estos nuevos materiales sobre diversas aplicaciones, ejemplificadas mediante el uso de líquidos iónicos funcionalizados con grupos quinona.
Palabras clave: líquidos iónicos de tarea específica; electroactividad; aplicaciones; dispositivos electroquímicos.
Introduction and perspective
Air and moisture stable room temperature ionic liquids (RTILs) were first reported by Wilkies and Zaworotko [1] in 1992. The materials reported were simple imidazolium salts of acetate ([Ac]) and tetrafluoroborate ([BF4]) as depicted in Fig. 1. This development represented a technical step-change from the earlier chloroaluminate molten salts [3] which were notoriously unstable under ambient conditions and thus opened up a vast array chemical and electrochemical possibilities which have been exploited ever since. The key physical or physiochemical properties that RTILs which make them attractive exhibit include
i) Inherent ionic conductivity (typically mS cm-1 range),
ii) inert / extreme redox robustness (up to 7 V potential window reported),
iii) non-volatile / non-flammable,
iv) high charge carrier concentration (>> 1 mol L-1),
v) thermal stability,
vi) liquid state at low temperatures.
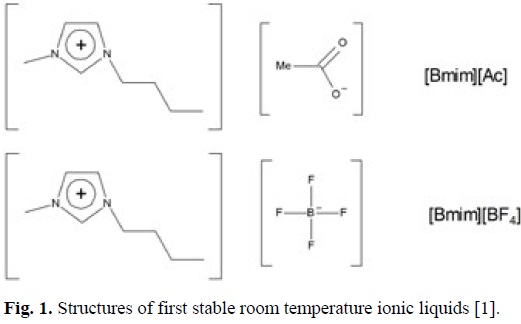
In addition to these favourable properties the idea of incorporating chemical functionality into the RTIL structure to create task-specific ionic liquids (TSILs) [4, 5] has contributed greatly to the development of unique applications beyond their use as simple electrolytes or solvents. Although Davis' 2004 paper [5] formally introduced TSILs, examples of ILs with built-in electro-active functionality were already known in the literature; for example, Murray et al. incorporated both quinone [6] and metal complexes [7] into "molten salt" structures while viologen IL materials were reported in 2003 [8]. Contemporaneous to Davis, other work by Shreeve et al. [9] and Park et al. [10] concentrated on the incorporating of ferrocene (Fc) moieties (as functionalise cations) and Γ (as anions), respectively. These works appeared in 2004 while Wu et al. [11] subsequently reported the incorporation of the redox catalyst TEMPO (2,2,<5,<5-tetramethylpiperidine-1-oxyl/) into imidazole-bases salts which were solids in their native state at RT but soluble in conventional ILs. More recently, Strehmel et al. [12] reported a TEMPO-based RTIL which acted as a radical probe in ILs.
It is probably not coincidental that these early examples emerged from classical molecular electrochemistry and that they all point in the direction of important technical applications such as enzyme redox catalysis (Fc/Fc+), dye sensitizes solar cells (I-/I3-) (DSSCs), hydrogen peroxide generation (qui-none/hydroquinone) and alcohol oxidations (TEMPO/nitrosonium). Presenting these and other well-known electrochemistries in appropriate IL form will lead to new or improved electrochemical technologies including the aforementioned but also including redox flow batteries, thermal batteries, and super-capacitors. Since most of these applications involve the harvesting and energy storage functions their emergence and exploitation may impact significantly on the energy landscape in the foreseeable future.
Students of molecular electrochemistry and redox catalysis can easily trace the evolution of the field from homogeneous-bases (i.e. solution-based) processes to heterogenised systems with the advent of chemically modified electrodes in the early 1980s [13], in particular, for redox catalytic systems based on surface confined redox centres (2-D) or for polymer-confined (3-D) systems. In principle, heterogenising these chemistries obviated, or simplified, the recovery of valuable redox catalysts from complex reaction liquor, or facilitated the construction of selective thin-film or thin-layer electrochemical devices such as sensors or DSSCs.
Because ILs can be designed to facilitate product recovery [4] and are inherently non-volatile, redox active ionic liquids can be viewed as a further evolution in molecular electrochemistry / redox catalysis / redox mediators despite presenting themselves as bulk liquids. Indeed, bulk liquids have the advantage of scaling by volume rather that by area in synthetic and energy storage applications so are valuable per se. Also, in terms of thin film type applications (e.g. sensor) Toniolo et al. [14] have already successfully demonstrated an O2 electrochemical sensor deploying an anthraquinone based redox catalytic ionic liquid.
A third possibility for deployment of redox active ionic liquids is as a "sandwich" device i.e. positioned between two active electrodes e.g. as electron transfer mediators in DSSCs or as "redox-active electrolytes" in capacitors. For the latter, such systems would act as supercapacitors since they will harness the faradic charge stored in the built-in redox centres as well as the electrostatic capacitive charge [15]. For the former I- based ionic liquids have been reported [10].
Although ionic liquids exhibit many attractive properties a potentially serious limitation of these materials is their relatively large viscosities which typically range from ca. 20 cP to 1000s of cP with the common imidazolium and pyrrolidinium ILs (depending on the anion) exhibiting viscosities in the 50 -350 cP range. Significantly, Gratzel et al. have shown that ion conductivity (S cm-1) in RTILs is an inverse function of viscosity [16] so for all electrochemical devices which rely on mass transport minimising viscosity is critical. Since viscosity is intrinsically dependent on the complexity (size and structural symmetry) of the cation and the nature of the anion (e.g. H-bonding ability) the design of new functional TSILs requires appropriate careful consideration. From an intuitive empirical perspective low viscosity is achieved with small non-coordinating anions paired with larger, but not too large, structurally asymmetric cations where charge delocalisation is extensive for both ions. Even for simple RTILs, they only exist as liquids over a fairly narrow range of structures. Obviously, adding functionality to RTILs inevitably increases mass, and frequently increases structural symmetry and inter-molecular attractive forces all of which favour solidification; these restrictions imposes severe challenges to molecular design of TSILs.
In addition to minimising viscosity the following functional requirements must also be met,
• bear a permanent charge i.e. be a RTIL in both redox states,
• exhibit reversible electrochemistry,
• be catalytic i.e. appropriate E0 for redox catalysis,
• be otherwise inert,
• be either hydrophobic or hydrophilic as application requires.
Although there are many well-known redox chemistries to choose from the syntheses, characterisation and electrochemistry of some novel quinone-based ionic liquids will be presented and discussed here in terms of their potential for application in real electrochemical devices and processes, in particular as O2 reduction redox catalysts. The core quinone structure is shown in Fig. 2.
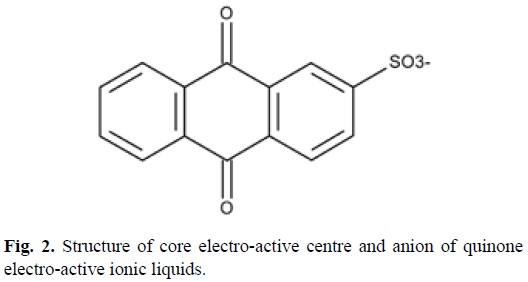
Experimental
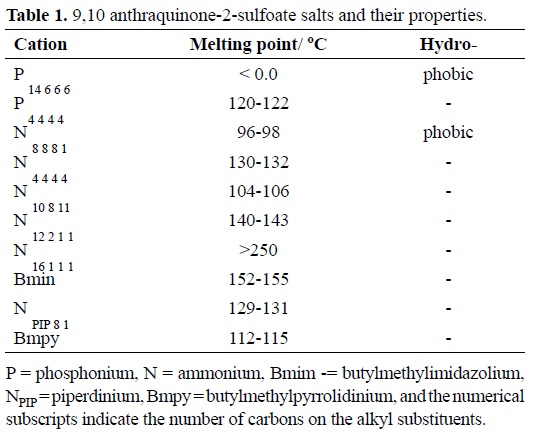
Synthesis of 9,10-anthraquinone-2-sulfonate functionalized ionic liquids. Sodium anthraquinone-2-sulfonate ([Na][AQS]) was obtained from Aldrich. The sulfonate anion provided the common anion for a suite of familiar quaternary ammonium and phosphonium ionic liquid cations (Table 1). A series of chloride salt of these cations were reacted with sodium anthraqui-none-2-sulfonate in toluene for 48 Hrs. with constant stirring after which the NaCl precipitate was removed by filtration (No. 4 sintered glass filter). The resultant liquor was washed repeatedly with deionised water to remove residual NaCl. The toluene was removed under vacuum to reveal the salt product. Details of their melting points and hydrophobicity are given in Table 1.
Electrochemical Measurements. All electrochemical measurements were performed using a three-electrode potentistat, either a Voltalab 40 (Radiometer) or a Digi Ivy 2100 mini-potentiostat. The working electrode in all experiments were 3mm diameter glassy carbon (from Bioanalytical Systems (BAS)) while the counter electrodes were Pt wire (Goodfellow) sealed in glass. The working electrodes were polished with 0.05 |im y-alumina prior to use. The reference electrodes were either aqueous Ag/Ag+ (from BAS, 1.0 M AgNO3), a saturated calomel electrode (SCE) or a Pt wire quasi-reference electrode (Ptss). Details of electrolytes and solvents used are given in the text and/or in Figure legends.
Results and Discussion
Preliminary observations: The accepted definition of RTILs is salts which have melting points ≤100 oC. Despite this generous technical definition practical considerations desire, or demand, much lower melting points, for example to facilitate enzyme electrochemistry or to prevent accidental solidification of the functional liquid in reactors during shut-down etc. Inspection of the data in Table 1 indicates that only one of the ten anthra-quinone-2-sulfonate is liquid at 293 K and that only two out of these ten fall within the accepted upper melting point limit defining RTILs. These observations are not surprising since the symmetry and planarity of the AQS core, coupled with the significant pi-pi inter-molecular interaction between quinone molecules increases the likelihood of long-range order so solidification rather than liquefaction is likely.
Electrochemistry of Anthraquinone-2-sulfonates: It is our experience that the common cation does not interfere with the electrochemistry of the quinone-bases salts therefore only the electrochemistry of [P14 6 6 6][AQS] will be presented here. Fig. 3 shows cyclic voltammograms for [P14 6 6 6][AQS] in O2-free acetonitrile containing 0.2 mol L-1 tetrabutylammonium tetra-fluoroborate (TBATFB) electrolyte recorded with a Digi Ivy mini potentiostat (iR compensation function unavailable). It is clear that the AQS ion undergoes two sequential and reversible electrochemical reductions via the classical two one-electron events as expected (Equations 1 and 2) resulting in the formation of stable radical anions and stable di-anion, respectively.
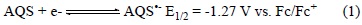
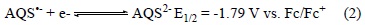
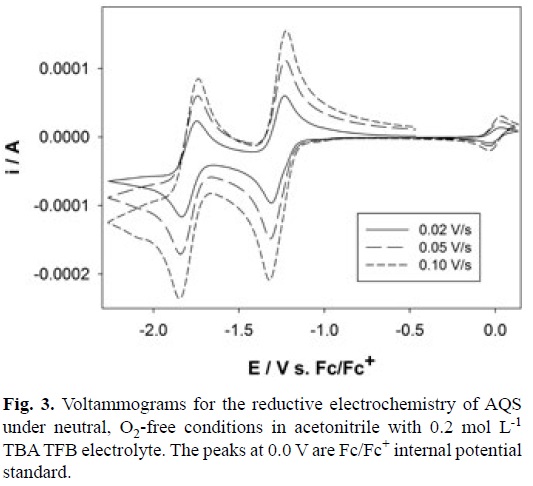
Using the Randles-Sevcik equation the diffusion coefficients (D) for both AQS and AQS'- were determined to be 1.1 x 10-5 ± 0.1 x 10-6 cm2 s-1 and 1.4 x 10-5 ± 0.3 x 10-6 cm s-1, respectively. A "worst case scenario" estimation of k0, the rate of heterogeneous electron transfer, for both redox processes calculated from peak-to-peak separations (ΔEp), is ~6 x 10-3 and ~5 x10-3 cm s-1 for the first and second redox processes, respectively.
Transferring the electrochemistry to the neutral ionic liquid environment of [P14 6 6 6] [NTf2] recorded with iR compensation using a Voltalab 40 potentiostat has no effect on the general electrochemistry insofar as the mechanism is the same and both the radical anions and dianions are stable as can be seen in Fig. 4.
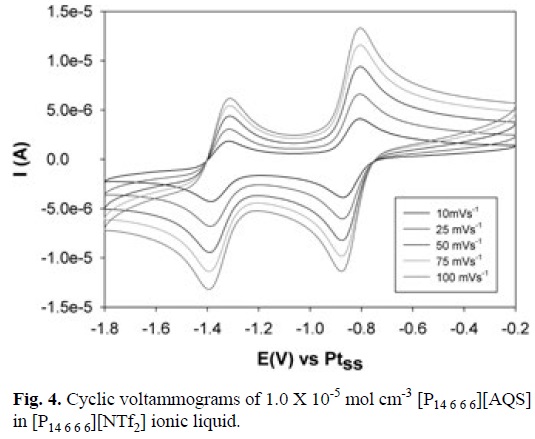
It is also evident from the data in Fig. 4 that due to the viscous nature of the ionic liquid the rate of mass transport is significantly reduced relative to acetonitrile where a diminution of the [AQS] diffusion coefficient from —10-5 cm s-1 to 5 x 10-8 cm s-1 is evident. On the positive side, ko for both process is ≥ 0.1 cm s-1, a rate which is considered electrochemically reversible i.e. fast.
The electrochemical generation of AQS'- was used previously by Toniolo et al. [14] to develop an ionic liquid based thin-layer O2 sensor where the sensing redox catalytic reaction was the formation of super-oxide according to Equation 3.
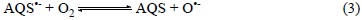
Two unique features of this sensor are both worth considering; firstly, since the RTILs are non-volatility the sensor never "dries out", and secondly, the thin-film of electro-active RTIL is easily replaced to renew the sensing interface.
It is well known that quinone electrochemistry is pH-de-pendent therefore the electrochemistry of AQS under acidic conditions (0.2 mol L-1 HClO4) in acetonitrile is shown in Fig. 5. It is immediately obvious from the traces in Fig. 5 that the reduction involves a single process while the re-oxidation potential is significantly displaced anodically from the expected reversible potential. In addition, the anodic and cathodic peak potentials migrate positively and negatively with increasing potential sweep-rate, respectively. This behaviour is the classical electrochemistry for quinones under the influence of complex acid-base equilibrium processes coupled with electron transfer [17]. What is clear from Fig. 5 is that the overall electrochemical reduction process under acidic conditions is a single process occurring at ca. 0.2 V vs Ag/Ag+ involving two electrons and two proton i.e. electrochemical hydrogenation to form the anthrahydroquinone species (AH2QS) according to Equation 4. The literature value for the reversible AQS/AH2QS process is 0.225 vs SHE [18].
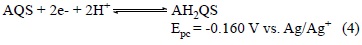
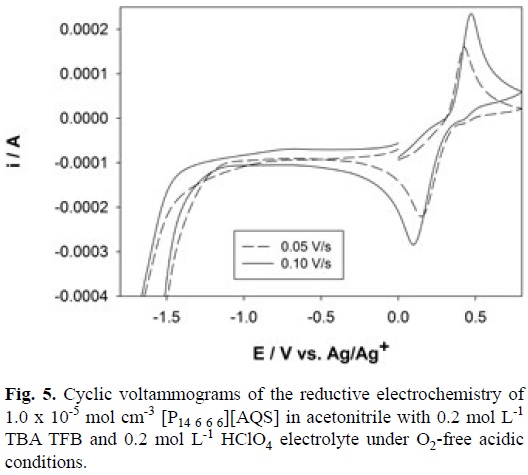
AQS electrochemistry in [P14 6 6 6][NTf2] acidified with three stoichiometric equivalents (per carbonyl) of the super-acid HNTf2 is shown in Fig. 6 where it is clear that the reduction occurs in a chemically reversible fashion but ΔEp (peak-to-peak separation) of up to 500 mV suggest significant influence of coupled proton transfer equilibria at play within the non-aqueous IL environment. Overall, the electrochemistry occurs via the expected 2-electron/2-proton mechanism leading to the redox-catalytic hydroquinone form.
It is well known that anthrahydroquinones are very useful reducing agents with O2 reduction to H2O2 being used commercially to produce millions of tonnes of peroxide annually. This second-order [19] chemical process is known as auto-oxidation and exploits H2 rather than electricity as the energy source. In order to assess the reactivity of the electro-generated AH2QS towards O2 reduction oxygen gas was introduced into the acidic AQS solution (equilibrated at 1 atm. O2 atmosphere) and slow-sweep-rate cyclic voltammetry recorded as depicted in Fig. 7 where a Sigmoidal curve is observed due to the steady-state catalytic current, ic, behaviour which is indicative of redox-catalysis occurring at the electrode according to Equations 4 and 5.
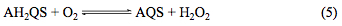
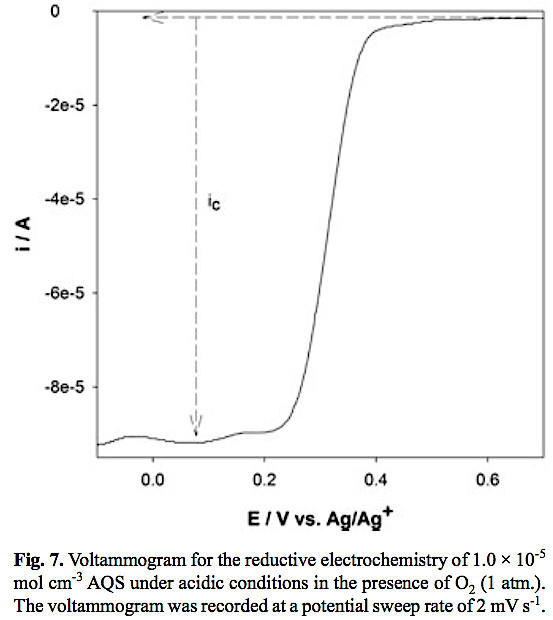
Using Equation 6, where n is the number of electrons (n = 2), F is Faraday's constant, A is the area of the electrode and C is the concentration of AQS (1.0 x 10-5 mol cm-3) a forward rate constant (kf, s-1) can be calculated assuming pseudo-first order kinetics ([O2] >> [[AQS]] since the solubility of O2 in pure acetonitrile is 8.1 mol L-1 [20] and ~1 mol L-1 in the presence of perchlorate electrolyte [21].

For the highly acidic solution (0.2 mol L-1 HClO4, = pH 0.7 assuming full dissociation) with saturated [O2], kf was found to be 0.03 s-1 which corresponds to a 2nd order rate constant of 3.0 mol-1 L s-1. Although relatively slow, this rate is somewhat faster than the rate of AQ hydrogenation using H2 in catalytic slurry reactors e.g. — 0.02 mol m-3 s-1 [22]. Because the E0 for AQS AH2QS decreases by 58 mV / pH unit [23] and that the O2 reduction reaction is pH-dependent through proton transfer steps (Equation 4), the rate of O2 reduction reaction will increase logarithmically with pH as previously found for redox catalytic alcohol oxidation reactions in ionic liquids [24].
Conclusions
Based on the simple examples shown here it is evident that building electrochemical functionality into ionic liquid "type" structure is relatively straightforward from a synthetic perspective but the likelihood of success (obtaining a RTIL) depends entirely on the nature of the anion and cation, therefore restricting synthesis to commercially available ions will inevitably severely limit the functional materials obtained. Out of the ten examples shown here it is no coincident that P14 6 6 6 exemplar "works" because it, even as the chloride salt, behaves effectively as an oil rather that as solid salt. In terms of deploying these types of materials, whether RTIL or not, it should be kept in mind that they are soluble/miscible in RTILs with a common counter-ion e.g. in low viscosity carrier ILs such as [Bmim] [NTf2] and thus the "business end" of the functional salt can be deployed in RTIL form. It is also worth keeping in mind that it is the nature of that anion (usually non-coordinating) that predominantly controls the physical properties of RTILs so switching the permanent charged on the quinone to cationic present an opportunity to create redox active quinone-based RTILs with more favourable physical properties. Incorporating redox behaviour in this way for redox catalysis and redox conduction creates possibilities for new electrochemical processes and devices [25].
Acknowledgments
L. G. would like to thank the Department of Education and Learning in Northern Ireland for a PhD Studentship. L. D thanks QUB for a PhD studentship. APD and G.G.W thanks. The Royal Society of London for an International Collaboration Grant.
References
1. Wilkes, J. S., Zaworotko, M. J. Chem. Commun. 1992, 13, 965-967. [ Links ]
2. Hurley, F. H., Wier, Jr., T. P., US Patent 2,446,349 (1948). [ Links ]
3. Chum, H. L., Koch, V. R., Miller, L. L., Osteryoung, R. A. J. Am. Chem. Soc. 1975, 91, 3264-3265. [ Links ]
4. Welton,T. Chem. Rev. 1999, 99, 2071-2083. [ Links ]
5. Davis, J. H. Chem Lett. 2004, 33, 1072-1077. [ Links ]
6. Williams, M. E., Murray, R.W. J. Phys Chem. B. 1999, 103, 10221-10227. [ Links ]
7. Masui, H, Murray, R. W. Inorg. Chem. 1997, 36, 5118-5126. [ Links ]
8. Bhowmik, P. K., Haesook, H, Cebe, J. J., Burchett, R.A. Liquid Crystals, 2003, 30, 1433-1440. [ Links ]
9. Gao, Y., Twamley, B., Shreeve, J. M. Inorg. Chem. 2004, 43, 2406-3412. [ Links ]
10. Kang, M. G. Ryu, K. S. Chanh, S. H., Park, N. G. ETRI 2004, 26, 647-652. [ Links ]
11. Wu, X. E., Ma, L., Ding, M.X., Gao, L.X. Synlett. 2005, 4, 607-610. [ Links ]
12. Strehmel, V., Rexhausen, H., Strauch, P. Tet. Lett. 2008, 49, 7143-7145. [ Links ]
13. Murray, R.W. Electroanalytical Chemistry, 1984, 13, 191-368. [ Links ]
14. Toniolo, R., Dossi, N., Pizzariello, A. , Doherty, A .P. , Susmel, S., Bontempelli, G. J. Electroanalytical Chem. 2012, 670, 23-29 [ Links ]
15. Roldan, S., Blanco, C., Granda, M., Menendez, R., Santamaria, R. Angewendte Int. Ed., 2011, 50, 1699-1701. [ Links ]
16. Bonhote, P., Dias, A. P., Papageorgiou, N., Kalyanasundaram, K., Gratzel, M., Inorg. Chem, 1996, 35, 1168-1178. [ Links ]
17. DuVall, S. H., McCreery, R. L. J. Am. Chem. Soc, 2000, 122, 6759-6764. [ Links ]
18. Bailey, S. I., Ritchie, I. M. Electrochim Acta, 1985, 30, 3-12. [ Links ]
19. Levenspi, O, Godfrey, J. H., Chem. Eng. Sci., 1974, 29, 1723-1730. [ Links ]
20. Laoire, C. O.,Mukerjee,S., Abraham, K. M., Plichta, E. J., Hendrickson, M. A. J. Phys. Chem. C, 2010, 114, 9178-9186. [ Links ]
21. Li, Q, Batchelor-McAuley,C., Lawrence, N. S., Hartshorne, R. S., Compton, R. G., J. Electroanal. Chem, 2013, 688, 328-335. [ Links ]
22. Fayyaz Khan, M F., Ramzan, O., Mukhtar, A., Shafiq, U., Khan, A. F. Res. J. of Chem. Sci., 2015, 5, 48-52. [ Links ]
23. Ojani, R., Raoof, J., Ebrahimi, M., Iran J. Chem. & Chem. Eng. 2001, 20, 75-82. [ Links ]
24. Barhdadi, R.,Troupel, M., Comminges, C., Laurent, M., Doherty, A. P. J. Phys. Chem. B. 2012, 116, 277-282. [ Links ]
25. Murray, R.W. J. Electrochem. Soc. 1984, 132, 833-839. [ Links ]

