










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.59 no.4 Ciudad de México oct./dic. 2015
Article
Free Radicals Induced Oxidative Stress at a Molecular Level: The Current Status, Challenges and Perspectives of Computational Chemistry Based Protocols
Annia Galano1
1 Departamento de Química. Universidad Autónoma Metropolitana-Iztapalapa. San Rafael Atlixco 186, Col. Vicentina. Iztapalapa. C. P. 09340. México D. F. México. To whom correspondence should be addressed. E-mail: agalano@prodigy.net.mx; agal@xanum.uam.mx
Received September 14th, 2015;
Accepted January 18th, 2016.
Abstract
Oxidative stress is frequently caused by an excess of free radicals and has been associated with a wide variety of health disorders. Therefore, finding strategies for scavenging free radicals has become an active area of research. This review summarizes, from a physicochemical perspective, relevant strategies to fight oxidative stress via antioxidants, including prevention, deactivation of oxidants, and repair of damaged targets. Different reaction mechanisms involved in the chemical protection exerted by antioxidants are discussed, as well as their relative importance depending on several aspects. Some of them are the polarity of the environment, the pH of aqueous phase, and the chemical nature of the reacting radicals. Data that can currently be obtained from computational, quantum, chemistry, protocols are detailed and their reliability is analyzed. Viable criteria to identify optimal antioxidants using such protocols are provided. Current challenges and future directions in this area of research are discussed. A large set of antioxidants are compared and their trends in activity, based on kinetic data, is provided.
Key words: antioxidants; free radical scavenging; kinetics; mechanism of reaction; trends in activity.
Resumen
El estrés oxidativo frecuentemente es causado por un exceso de radicales libres, y ha sido asociado con una amplia variedad de problemas de salud. Es por ello que encontrar estrategias viables para eliminar radicales libres se ha convertido en una activa área de investigación. Esta reseña resume, desde una perspectiva fisicoquímica, estrategias relevantes para combatir el estrés oxidativo por medio de antioxidantes incluyendo prevención, desactivación de oxidantes, y reparación de blancos dañados. Se discuten diferentes mecanismos de reacción involucrados en la protección química que ejercen los antioxidantes, así como su importancia relativa dependiendo de diferentes aspectos. Algunos de ellos son la polaridad del ambiente, el pH en solución acuosa, y la naturaleza química de los radicales libres. Se detalla la información que puede obtenerse actualmente a partir de protocolos basados en la química computacional y se analiza su confiabilidad. Se proporcionan criterios viables para identificar antioxidantes óptimos, usando estos protocolos. Se discuten algunos de los retos actuales y de las perspectivas futuras en esta área de investigación. Un amplio conjunto de antioxidantes son comparados y se propone su tendencia en actividad, en base a datos cinéticos.
Palabras clave: antioxidantes; desactivación de radicales libres; cinética; mecanismos de reacción; tendencias de actividad.
1. Introduction
Saying that we all want to live long might seems to be a trivial statement. However a long lifespan is not our only goal. We also want to have a high quality of life, which necessary involves maintaining a good health status. This is, beyond any doubts, quite a challenge. Oxidative stress (OS) is one of the most important factor threatening this aspiration. It has been demonstrated to be involved in numerous and diverse health disorders, as well as in some deleterious effects of aging. Therefore, it is not surprising that understanding the damages caused by OS, and finding efficient strategies to reduce it, have become active areas of research. In fact, the number of publications on both topics have significantly increased in the last two decades (Fig. 1). In addition, it seems interesting to note that both lines of investigation have almost parallel trends, thus providing the necessary mutual feedback.
OS can be considered a chemical process, but it is a very complex one. It takes place under varying conditions, involving a wide variety of chemical species and competing reactions. This complexity makes OS related investigations particularly difficult. In vivo studies have the ultimate answers, but they mainly deal with OS from a phenomenological approach, i.e. with the effects of triggering and ameliorating factors, and the associated responses. The chemical details on OS are usually not acquired this way. On the other hand, in vitro and in silico studies are able of providing such information but they are necessary based on simplified models of the actual processes taking place within living organisms. Comprehending in full detail the chemical damage caused by OS to biomolecules, the chemical processes involved in its prevention, and the global effects in living systems, are all crucial aspects for designing efficient strategies against OS. Thus, as in many other fields of science, it seems that multidisciplinary approaches are essential in the investigation of OS since simultaneously analyzing the information gathered from different kinds of investigation seems to be the only way of obtaining a whole picture of this complex phenomenon, and of envisaging potential solutions.
In this review, OS is analyzed from a molecular point of view, mainly focused on the currently available computational tools. Quantum mechanics based studies are currently considered important approaches for addressing specific chemical problems. They constitute viable alternatives to experiments, especially when experimental studies are particularly difficult, expensive, or even impracticable. Moreover, they usually provide complementary information to that obtained from experimental approaches, frequently leading to successful multi-disciplinary investigations. At the same time, the value of using theoretical approaches is ruled by the accuracy of the obtained results. Fortunately, nowadays it is possible to obtain reliable data from calculations, at practical computational costs, for systems of relatively large sizes since computational power has increased, spectacularly, in the last decades. Therefore, computational strategies have become an appealing option to investigate OS related chemical processes.
2. Oxidative Stress
OS arises as a consequence of a chemical imbalance between the production and consumption of oxidants within biological systems.[1] Free radicals (FR) are among such oxidants. They are not intrinsically dangerous, but as it is the case for almost anything in life, they can be harmful or beneficial, depending on their amounts. Living organisms are designed to maintain a balance between FR production and removal, which is intended to keep FR at low to moderate concentrations. Under such conditions these chemical species are essential to optimal human health. They are involved in several biological processes including mitogenic responses[2-5] and maturation of cellular structures. [6] FR also have roles in the defense[7, 8] and cellular signaling[3, 5, 9] systems, as well as in the apoptosis of defective cells [10, 11] and in the regulation of insulin receptor kinase activity.[7]
On the contrary, at high concentrations, FR are toxic to living organisms. But, if living organisms are designed to properly deal with FR production, what may cause them to reach unhealthy concentrations? The problem arises from the fact that they are not only produced endogenously but also exogenously. In both cases there is a vast number of sources contributing to increase FR amounts to such extent that only a fraction of them are consumed through the physiological process intended to do so. Endogenous FR are generated from inflammation, immune responses, ischemia, infection, mental or physical stress, and aging.[12-22] Exogenous FR arise from environmental pollution, heavy or transition metals, cigarette smoke, certain drugs, alcohol, and radiation.[23-36] Thus, considering the abundant number of FR sources that we are exposed to in the modern world, keep FR at healthy concentrations is currently a challenge.
While the best way to prevent OS, and the associated health risks, is logically avoiding exposure to FR -and other oxidants- sources this strategy is far from being easily achieved. Fortunately, FR concentrations can be diminished using alternative chemical ways to remove them, for example increasing our intake of antioxidants.
2.1. Free Radicals, Chemical Features and Reactivity
Free radicals are characterized for containing one or more unpaired electrons. This feature makes them particularly reactive, and is also responsible for the FR ability to trigger chain reaction mechanisms, propagating the associated molecular damage. A wide variety of FR can be found in living systems. Most of them are, or arise from, reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive sulfur species (RSS). ROS include oxygen-based free radicals, such as the superoxide radical anion (O2'-), hydroxyl ('OH), alkoxyl (RO'), organic peroxyl (ROO') and hydroperoxyl (HOO') radicals. RNS comprise peroxynitrite (ONOO-), nitric oxide (NO') and nitrogen dioxide (NO2'), while the most common RSS are thiyl radicals (RS'), sulfenic acids (RSOH), and disulfide-S-oxides (RS(O)2SR).
Regarding their reactivity, 'OH is the most reactive and dangerous species among ROS, thus it will be further discussed in more detail in section 2.3.1. ROO' are significantly less reactive species, which allow them to diffuse to remote cellular locations,[37] having half-lives in the order of seconds.[38] RO' are formed from the reduction of peroxides and are significantly more reactive than ROO', provided that R is the same in both species, while they are less reactive than 'OH.[39-43] Concerning RNS, the chemical reactivity of NO' is rather low, and therefore its direct toxicity is actually minor.[44, 45] On the other hand, it reacts with O2'- yielding peroxynitrite,[46] which is a potent oxidant and a very damaging species able to react with lipids, proteins, and DNA.[47-49] Nitrogen dioxide is a moderate oxidant, and its reactivity is between those of NO' and ONOO-. NO2 reacts with organic molecules at rates that range from ~104 to 106 M-1 s-1, depending on the pH.[50, 51].
RSS are believed to be mainly formed as products of the reactions of thiols with ROS and RNS,[52] thus they are expected to be less reactive than their parent O and N species. However, they are still able of damaging proteins.[53-55] Within this context it seems relevant to mention the investigations performed by Asmus group, who have gathered relevant information about the sulfur 2-center-3-electron bonded radical species (2c3e-S.\S). Some examples of these species are RSSR'-, RSSR'+, and R2SX with X=halide.[56-60] RSSR'-constitutes and interesting case since it is in equilibrium with the corresponding thiyl radical. However, while RSSR'- is a reductant that may react with O2 yielding O2'-, RS' is a moderate oxidant.[56]
Because of their important roles in electron transfer reactions within biological environments, several theoretical studies have been devoted to provide information on 2-center-3-elec-tron bonded species. Albeit this subject alone would deserve a full review, some representative examples are provided here, since they are nice cases where theory and experiments feedback has contribute to a better understanding of biologically relevant species. The interested reader can find more information on this subject elsewhere.[61]
In a very early study, McKee performed a theoretical investigation on the bond strength and configuration of 2c3e-S∴S for a series of charged acyclic dithiols, HS(CH2)nSH+ (with n = 1-4).[62] In this work it was found that the bond strength increases with n, except for the n = 3 which is slightly more stable than the bridged ion with n = 4, in agreement with the experimental data. In addition, the properties of intramolecular 2c3e bonds were rationalized as a compromise between maximizing orbital overlap and minimizing steric repulsion.
More recently, Brunelle and Rauk performed a theoretical investigation on the effect of three-electron bonding on the reduction potential of the radical cation yielded by one-electron oxidation of methionine residues (Met'+), in peptide environments.[63] They proposed that Met'+ stabilization by three-electron bonding is feasible when an S∴N bond can be formed with a free amino group, for example in an N-terminal Met or a neighboring lysine. In such cases a substantial lowering of the reduction potential was predicted, with implications for the re-dox chemistry associated with Alzheimer's disease. These findings are in line with the experimental results reported by Schõneich et al.[64] In addition, last year Wiberg and Petersson performed a systematic investigation on the bond dissociation enthalpies (BDE) for a series of RX-H compounds with X = CH2, NH, O, PH, and S.[65] They related most of the substituent effects to a conjugative interaction in the 2c3e radicals formed by H abstraction. The good agreement between their theoretical results and the experimental BDE values, supports this interpretation.
2.2. Damage
The toxicity of FR was first reported about 60 years ago by Gerschman and coworkers,[66] who proposed that these species are responsible for the damaging effects of oxygen poisoning and ionizing radiation. Despite of the important implications of this discovery, it remained almost ignored for a long time. Nowadays there are numerous reports supporting this finding and providing evidence on the role of OS, and excess of FR, in the onset and development of a large number of health disorders. OS has been associated with pulmonary,[67-78] renal,[7, 79-86] and ocular[87-93] diseases; rheumatoid arthritis,[94-98] as well as with pre-eclampsia and fetal growth restriction.[99-104] The development of some kinds of cancer has also been associated with oxidative damage.[4, 5, 9, 105-113] It has been suggested that OS can be involved in several neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, memory loss, multiple sclerosis, and depression.[114-137] In addition, there is evidence indicating that OS may play a role in several cardiovascular diseases including congestive heart failure, atherosclerosis, ischemia, cardiomyopathy, cardiac hypertrophy, and hypertension.[7, 138-151] According to the overwhelming evidence connecting OS with numerous diseases, it is evident that finding efficient strategies to ameliorate OS is crucial to improve the human health status.
Regarding molecular damage, it has been proposed that one-electron oxidation reactions of DNA mainly involve guanine (G) sites,[152, 153] since it is the most easily oxidized of the nucleobases.[154-158] It seems important to call attention to the fact that despite of the small differences among the oxidation easiness of guanine, guanosine, 2'-deoxyguanosine, and 2'-deoxyguanosine 5'-monophosphate,[159] these compounds all have the lowest oxidation potential within the corresponding family. Consequently, the radical cation 2dG'+ is the most abundant one electron oxidized site in DNA. It can be formed through diverse oxidative processes including radiation, hole migrations from other nucleosides, and reactions with chemical oxidants.[160-169] 2dG'+ can further react, rapidly evolving into other species, through different reactions among which deprotonation is expected to be one of the most important due to the low pKa value of 2dG'+ (3.9).[170] It has been recently demonstrated that C centered radicals, in the sugar unit, are the main products yielded by the deprotonation of these oxidized sites at room, or body, temperatures.[171] These radical products are particularly dangerous because they may be involved in one of the most important type of DNA damage, the strand breaks.[172-177] Another product of the 'OH induced DNA oxidation is the 8-oxo-2dG radical adduct,[178-184] which has been used as a biomarker for oxidative stress.
The damaged induced by free radicals, particularly 'OH, to proteins can cause important structural modifications that eventually may lead to cross-link,[185-188] as well as spontaneous fragmentation, or increased proteolytic susceptibility.[189-194] Most amino acid residues have been identified as vulnerable to oxidative damage including cysteine,[195-202] histidine,[196, 203-205] methionine,[197-199, 202-208] tryptophan,[200, 202, 206] tyrosine,[198, 200, 205, 206, 208-210] asparagine,[211] leucine, lysine, serine, arginine, glutamine, and glutamic acid. [197] However some of them seem to be particularly susceptible to this kind of damage.
It has been demonstrated that sulfur-containing amino acid residues, methionine and cysteine, are particularly sensitive to the oxidation inflicted by almost all reactive oxygen species.[199, 212-214] Taking advantage of this behavior it has been proposed using cysteine supplementation to reduce DNA damage induced by sport training.[215] There is also evidence that in oxidized proteins and peptides there is a large amount of methionine sulfoxide, which is supposed to be produced through free radical intermediates.[216-222] This supports the high vulnerability of methionine residues to oxidative stress. In addition, it has been suggested that methionine residues may be involved in the free-radical-mediated oxidative stress of the amyloid β-peptide (Aβ), which has been associated with the Alzheimer's disease.[115-118, 223-231] In fact, it has been found that the removal of Met35, or its replacement by structurally similar amino acids such as norleucine (Nle), inhibits the aggregation of the Ap peptide and thus the related neurotoxic properties.[232-235] It has also been reported that methionine plays an important role on the oxidation of apolipoprotein D, which is up-regulated in Alzheimer's disease and upon oxidative stress.[218] It should be noted, however, that the relative reactivity of different amino acid residues towards free radicals can be significantly affected by surface exposure. This feature is expected to influence their oxidation kinetics, which may explain why some residues are more easily oxidized than others.[201]
Regarding the main sites involved in the oxidative damage to proteins and peptides, induced by free radicals, it has been proposed that both electronic and steric factors may play important roles on their relative rates. It has been found that the 'OH induced damage to a carbon sites in the backbone occur only for glycine and alanine, which has either no side-chain or only a methyl group. On the contrary, for residues with larger side-chains such as leucine or valine, the 'OH attack mainly involve side-chain sites.[236] It has also been proposed that the finding that reactions at sites other than α and β sites are the most favored ones can be explained by the influence of polar effects, structural factors, secondary interactions, and solvent effects, which have all been held responsible for variations in the reaction barriers. In addition it has also been suggested that the regioselectivity of hydrogen abstraction reactions from side-chains can also to be affected by hydrogen bonding to, or protonation of, the substrate.[237] For radical adduct formation reactions, on the other hand, it has been found that 'OH additions to the different sites in the aromatic rings of tyrosine and phenylalanine are the most likely ones.[238]
2.3. Strategies to Reduce Oxidative Stress
There are different strategies that can be help to reduce OS. They all are intended to prevent, or minimize, the oxidative damage caused by FR -or other oxidants- to molecules of crucial biological importance such as DNA, proteins, and lipids. Depending on the moment at which they take place, they can be classified as prevention, protection or repairing strategies.
2.3.1. Prevention
OS prevention strategies refers to those actions that are taken to avoid oxidation by preventing oxidants from formation. The first way of achieving this is as simple as avoiding exposure to FR exogenous sources such as car exhaustion or chemically treated foods. However, as mentioned above, this is hardly ever possible. Another, more likely, way consists in inhibiting the endogenous production of oxidants. This can be achieved in several ways. For example, reducing exposure to UV-vis radiation, which is known to promote FR production particularly affecting exposed areas. In addition there are chemical processes that help inhibiting the formation of FR, in particular hy-droxyl radicals ('OH). This radical deserves particular attention because of its high reactivity, and the consequent widespread damage that it can cause. Among the oxygen-centered radicals, 'OH is the most reactive and electrophilic one.[239] In fact, its reactivity is so high that it is able of instantaneously attack almost any molecule in the vicinity of its site of formation. Its reactions with most chemical compounds occur at, or near to, diffusion-controlled rates (rate constants ≥ 108 M-1 s-1) with very low selectivity towards the different possible reaction sites. It has been estimated that this radical is responsible for about 60%-70% of the tissue damage arising from ionizing radiations,[240] and it has been held responsible for the most important oxidative damage to DNA.[241-243] Therefore inhibiting 'OH formation is expected to be an important way to reduce OS.
'OH can be produced by ultraviolet and ionizing radiations or from other radicals arising from enzymatic reactions. However, its main intracellular sources probably are the Fenton reaction and the metal catalyzed Haber-Weiss recombination (HWR). A formal distinction between these two reactions is made in here, albeit the Fenton reaction corresponds to the second step of the catalyzed HWR, for emphasizing on the fact that metal ions in different oxidation states are the initial reactants in each case and that their relative abundance in biological systems is quite different. The most likely metal ions that are involved in such processes are Fe and Cu.
The Fenton reaction, involves the reduced forms of these metals:
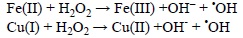
On the other hand, even though the Haber-Weiss recombination can be globally written as:
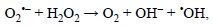
this reaction is too slow to be physiologically important, unless it is catalyzed by metal ions.[244] The catalyzed Haber-Weiss process becomes then a combination of two elementary chemical reactions. The first one involves the reaction of the superoxide radical anion (O2'-) with the oxidized forms of metal ions:
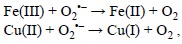
and the second step corresponds to the Fenton reaction.
There are two aspects of the Haber-Weiss recombination that are particularly important. The first one is that in the global process only O2'- and H2O2 are actually consumed while the metal ions act as true catalyst, i.e., they are regenerated during the overall process. Accordingly, a large amount of 'OH radicals can be produced from a very small number of metal ions. The second one is that the metal oxidized forms, i.e., Fe(III) and Cu(II), correspond to their most abundant and stable oxidative state. Therefore, in biological media it is expected that the relative importance of the first step of the HWR is higher than that of the direct Fenton reaction. However, it should be taken into account that Fe(III) and Cu(II) can also be reduced into Fe(II) and Cu(I) by other chemical species present in biological systems, such as the ascorbate ion. In any case, which seems to be important is that the reduction process -Fe(III) to Fe(II) or Cu(II) to Cu(I)- is the crucial step to the 'OH production. In other words, if the formation of the lower oxidation state ions, Fe(II) or Cu(I), is inhibited so is the 'OH production through the Fenton reaction, and therefore the 'OH-related oxidative damage. Accordingly, chelating agents able of decreasing the viability of Fe(III) and Cu(II) reduction reactions are expected to be effective for preventing, or inhibiting, oxidative stress.
Regarding the Fenton reaction, it is a complex process that in the above equations has been represented in a simplified manner. This process can be influenced by the pH, by the ligands bound to the metal ions, by the presence of other reductants and oxidants in the reaction environment, and also by enzymatic processes.[245-248] In addition, other metal ions with high oxidative power can be formed, such as the Fe(IV), as well as peroxo-complexes.[245, 249-251] Such complexes -as well as the hydrated metal ions- can bind to peptides, proteins, and other biological targets. For example it has been proposed that the iron-catalyzed oxidation of methionine in peptides, via the Fenton reaction, comprises two consecutive steps: (i) one-electron transfer reactions carried out by free, or complexed, hydroxyl radicals; and (ii) the reaction of an intermediary sulfur-nitrogen bonded radical cation with O2.[252] In the case of copper, there is experimental evidence supporting that some compounds can act as 'OH-inactivating ligand. They are supposed to protect against 'OH damage (i) by sequestering metal ions from reductants or (ii) by deactivating 'OH radicals as they are formed through Fenton-like reactions.[253] Accordingly, it is evident that investigating Fenton-related processes using computational tools is quite a challenge.
However, there are some recent examples illustrating the important information that can be gathered for these processes using computational chemistry tools. It has been proposed that after deprotonation, ellagic acid is capable of chelating copper in aqueous solution, yielding stable complexes.[254] These reactions were proposed to decrease the 'OH production, with larger concentrations leading to better protection. Thus, in addition to the ellagic acid free radical scavenging activity, metal chelation was suggested as an alternative way for this compound to exert its protection against OS. In another theoretical work, the copper sequestering ability of melatonin and its metabolites N1-acetyl-5-methoxykynuramine (AMK), N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), and cyclic 3-hydroxymelatonin (3OHM), was explored.[255] It was found that these compounds fully inhibit, via Cu(II) chelation, the oxidative stress induced by Cu(II)-ascorbate mixtures. In the same work melatonin, AFMK, and 3OHM were also proposed to be capable of turning off the first step of the HWR, thus fully preventing the 'OH production via the Fenton reaction. Two different complexation mechanisms were investigated, the direct-chelation mechanism and the coupled-deprotonation-chelation mechanism. The latter was found as the most likely one, under physiological conditions, based on thermochemical considerations. So it is proposed that the interaction with Cu induces deprotonation at the chelation site, which leads to particularly stable complexes. Based on the results from this study it was proposed that, concurrently with the previously reported free radical scavenging cascade, melatonin may also be involved in a "chelating cascade" contributing to reduce OS. Trends in reactivity suggested that, among melatonin and its metabolites, 3OHM is the most efficient for that purpose.
2.3.2. Chemical Protection
OS protection strategies refers to those actions that are taken to avoid oxidation by preventing oxidants from reaching biomolecules. One way to achieve this is by the presence of sacrifice targets, able of reacting with oxidants before they reach biomolecules. We are going to refer to these kind of molecules as antioxidants (a more detailed explanation on this concept is provided in the next section). For antioxidants to succeed in their protective action they must either be in higher concentrations, or react faster, than the molecules to protect. Since, in general the anti-oxidants concentrations are not higher than those of biomole-cules, under physiological conditions, a higher reactivity towards oxidants is the key factor for their protective effects. Therefore, kinetic analyses are expected to be particularly useful to investigate which compounds can be efficient as chemical protectors against oxidative stress. Moreover, some threshold value should be established to compared with, and thus allowing identification of those molecules able of reacting with oxidants faster than biomolecules. One possible criterion for that purpose is provided in section 5.3.1. Chemical protection is the main subject of this review and it will be discussed in detail in further sections.
2.3.3. Chemical Repairing
Unfortunately, prevention and protection are not always enough, i.e., the oxidative damage to biomolecules is not always avoidable. For example, as mentioned before 'OH radicals are so reactive that they are most likely to attack the molecule nearest to its production site, which might be a biological target such as DNA. Therefore, repairing the damaged sites before replication becomes crucial to maintain genomic integrity and a healthy status. Living organisms have defense mechanisms for such events, among which enzymatic repair has an essential role. In spite of this, it has been reported that enzymatic repairing systems have three major drawbacks.[256] First of all, the repairing enzymes are also susceptible to be damaged by OS, losing their function as a result of this damage. [257-262] In addition, when their action may be needed the most, i.e., during illness and aging, the enzymatic repairing activity is decreased.[263] Finally, but not less important, is the fact that the half-lives of DNA radicals are dramatically shorter than the enzymatic repairing processes. The first one is usually in the order of seconds,[264] while the second one can take hours.[265, 266] As a result, the protection exerted by enzymatic repair, against permanent DNA mutations, might not be enough. Fortunately OS-related damages can also be efficiently repaired by non-enzymatic, i.e., chemical, pathways.[256] They involve the fast removal of transient DNA radicals by natural and synthetic compounds.[256]
There are several chemical species that have been identified as viable candidates for that purpose, among which the most studied ones are polyphenols,[267-269] and singly substituted phenols.[270] Even though they may react through different mechanisms, it has been proposed that the DNA-radicals repairing processes by phenolic compounds are mainly governed by hydrogen transfer and single electron transfer reactions.[267, 268] It has also been proposed that during the repairing processes the electron transfer from phenols can take place combined with a proton transfer.[270-273] There are other chemical compounds that have also been reported to exhibit OS-damage repairing ability, including hydroxycinnamic acid derivatives,[271] indoles,[272] dopamine,[274] uric acid,[274] aniline,[275] and glutathione,[276, 277] which can also repair proteins.[278, 279]
Pellmar et al.[276] demonstrated that glutathione is crucial for repairing processes involving hippocampal neurons exposed to oxidative damage. On the other hand, Pujari et al.[277] provided evidence supporting that while glutathione does not act as a radio-protector against DNA damage induced by higher dose X-rays, it can modulate DNA repair activity. In a theoretical study exploring the repairing process of radical-damaged DNA by glutathione, the HT mechanism involving its thiol group was proposed as the most important route, being the main responsible for the repairing activity of this compound. [280] The rate constants for the repairing process were estimated to be close to the diffusion-limited regime. Accordingly, the reactions involved in such repair are fast enough for taking place before replication and thus for preventing the associated permanent DNA damage. Still this is another example where very intricate processes can compete or simultaneously take place. First, it has been proposed that the concentrations of glutathione found in tissues exposed to oxidative stress can be too low for efficiently eliminate thiyl radicals in peptides and proteins before they participate in other harmful processes.[281] At the same time, the product yield by the reactions of glutathione with ROS and other oxidants are thyl radicals themselves. Therefore they can, in turn, react with biological molecules. In fact S-glutathiolation is recognized as a result of the reactions of oxidants with proteins containing thiols.[282, 283] This process can alter the proteins functions, and it has been proposed that it may modify cell shape, signaling, ion transport, vascular tone, metabolism, mitochondrial function and transcription factors.[284]
3. Antioxidants
Antioxidants have been suggested to play important roles in the prevention of several chronic diseases.[110, 285] As a result, there are numerous works devoted to chemical compounds that exhibit antioxidant activity. However, the term antioxidant is often used in a rather loose way. For that reason it is important to clarify its meaning in the context of this review. Here, the term antioxidant refers to "any substance that when present at low concentrations compared to that of an oxidizable substrate would significantly delay or prevent oxidation of that substrate", which is the definition provided by Halliwel and co-workers.[286, 287] Within this definition the term oxidizable substrate refers to any biological target that is expected to be protected by the antioxidant, for example lipids, proteins, or DNA. In addition, due to the differences in their mechanisms of actions, it seems worthwhile to make distinctions between primary (Type I, or chain breaking) and secondary (Type II, or preventive) antioxidants. Albeit this classification has been proposed for lipid oxidation,[288] it can be extended to antioxidants protecting any other kind of biological targets.
Primary antioxidants are chemical species that prevent oxidation by acting as free radical scavengers. In other words, they directly react with free radicals, producing significantly less reactive species, or turning off the radical chain reaction. Secondary antioxidants, on the other hand, retard oxidation by indirect pathways which include metal chelation, decomposition of hydroperoxide to non-radical species, repair of primary antioxidants by hydrogen or electron donation, deactivation of singlet oxygen or sequestration of triplet oxygen, and absorption of ultraviolet radiation. In addition, some antioxidants can behave as multiple-function antioxidants, i.e., their protective effects are exerted by both primary and secondary ways of action.
3.1. Sources
Humans can obtain antioxidant from numerous sources, both produced within our bodies and acquired from food or diet supplements. Among the endogenous antioxidants there are the enzymatic ones such as the superoxide dismutase (SOD), and also non-enzymatic including melatonin, glutathione, coenzyme Q10, and lipoic acids. The exogenous sources can be classified in natural and synthetic depending on the way of production. Some natural exogenous antioxidants are polyphenols, carotenes, phenolic acids, ascorbic acid, vitamin E, etc; while some significant examples of synthetic antioxidants are gallates, N-acetilcistein and its amide, edaravone, butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), and ethoxyquin.
3.2. Characteristics of Ideal Antioxidants
Regardless of their sources there are several characteristics that are desirable for antioxidants. In fact, even though there are many molecules that exhibit antioxidant activity, not all of them are equally efficient for that purpose. A series of requirements have been proposed,[289] which allow identifying ideal antioxidants. They are:
- Toxicity: Obviously, this is the most important aspect to consider regarding the potential use of a compound as an antioxidant. It should be non-toxic before, and after, the antioxidant activity takes place. In addition, it is also important to be aware of possible interactions with any drug that may be concurrently consumed.
- Availability: Antioxidant should be available when needed. Therefore they should be easily acquired through the diet or produced in situ. As mentioned before, they can also be taken from dietary supplements. However, since OS is usually symptoms free, the latter is a more complicated way to assure consumption based on needs.
- Location and concentration: An efficient antioxidant should be not only ubiquitous, but also in adequate amounts in cells. This is because most free radicals have short half-lives within biological systems, due to their high reactivity. Accordingly, they are likely to react with molecules that are in the vicinity of their site of formation. Thus, antioxidants should be present in such sites at any time free radicals are produced in order to efficiently intercept them before reaching biological targets.
- Versatility: A good antioxidant should be able of easily reacting with different free radicals since there is actually a wide variety of them in biological systems. Then, ideally, an antioxidant should have the capacity of deactivating them all, as there is no way of predicting which free radical will find it first.
- Fast reactions: Based on the very definition of antioxidant, it becomes evident that for antioxidants to be able of efficiently protect biological targets they must react faster than the molecules to protect.
- Crossing physiological barriers: It is expected that a good antioxidant can be able of crossing physiologic barriers and to be rapidly transported into the cells, where they are needed the most. Therefore amphiphilic molecules, i.e., those with both hydrophilic and lipophilic character, are particularly desirable. In addition, their size is also important since it should be optimum for transportation across cellular membranes.
- Regeneration: In this context the term regeneration refers to antioxidants that are able of scavenging several radical equivalents. Antioxidants that have physiologically mechanisms that regenerate their original form are expected to be particular efficient for reducing OS, since they would be capable of scavenging more than one free radical. In addition albeit reactions between antioxidants and free radicals yield oxidized forms of the antioxidants that have -by definition- less scavenging activity than the original compound, in some cases such oxidized species can still efficiently deactivate free radicals.
- Minimal loss: To avoid large urinary losses that can cause short half-lives, ideal antioxidants should be suitable to be reabsorpted after filtered by the kidneys. In addition, the concentration of any chemical compound is reduced in physiological environments by metabolic routes. Therefore, those antioxidants with metabolites that still present antioxidant activity are expected to be particularly efficient, for example melatonin.
4. Reaction Mechanisms
The reactions involved in the antioxidant activity of chemical compounds take place in very complex environments. This complexity arises from the large numbers of species present in biological media that may be involved in simultaneous, and competing, chemical reactions. Their relative importance would depend on both their concentration and intrinsic reactivity. In addition, chain reaction mechanisms may also be involved because of the very chemical nature of free radicals. Accordingly, subsequent chemical processes can rapidly follow the first oxidation step. In this regard, it is also important to note that different radicals not necessarily react via the same mechanism, and that the polarity of the environment, as well as the pH in the aqueous phase, can also alter the relative importance of the competing reactions. Therefore it becomes evident that elucidating the main reaction mechanisms involved in the antioxidant activities of chemical compounds may be a challenge.
Both experimental and theoretical approaches can be used to address this difficult task. From an experimental point of view, a good strategy may be to perform detailed product analyses. However, this approach may involve a rather large degree of inference because the processes usually take place at high rates and comprises several, parallel or consecutive, steps. Therefore, the observable products are often mixtures yielded by several elementary reactions. In addition, the same products may be produced through different mechanisms. Computational strategies also involve numerous difficulties. They are mainly related to the inevitable use of simplified models, and also to the availability of reliable strategies for properly including environmental factors such as solvent effects. That is why, the best way to address this important part of the antioxidant activity is probably by combining experimental and theoretical efforts.
Some of the most important reaction mechanism involved in antioxidant protection are revised in this section, with the intention of separately analyze the possible chemical routes contributing to the observable, overall, antioxidant activity of chemical compounds.
4.1. Single Step Mechanisms
4.1.1. Radical Adduct Formation (RAF)
The potential role of this mechanism is ruled by the antioxidant structure, in particular by the presence of multiple bonds. The nature of the FR also have an effect on its viability. In general electrophilic FR are the most likely to be involved in RAF reactions. In addition the reaction site should be exposed, and the size of the FR should be from small to medium to avoid important steric effects that may prevent RAF reactions from taking place. The RAF mechanism can be schematically represented as:
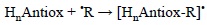
where HnAntiox and 'R are the antioxidant and the free radical, respectively.
There are several examples of antioxidants that are prompt to react via RAF. For example, it has been proposed that this mechanism is particularly important for carotenoids when reacting with the following radicals: 'OOH,[290] glutathione and 2-mercaptoethanol thiyl,[291] alkyl, alkoxyl, and alkylperoxyl,[292] and benzylperoxyl.[293] RAF has also been proposed to be a significant mechanism for the 'OH scavenging activity of gentisic acid,[294] caffeine,[39] edaravone in non-polar solvents,[295, 296] melatonin,[297] and its metabolites AMK, AFMK and 3OHM,[298, 299] hydroxybenzyl alcohols,[300] rebamipide,[301] and carnosine.[302]
4.1.2. Single Electron Transfer (SET)
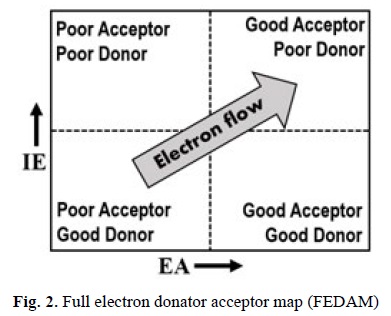
The viability of this mechanism is usually ruled by the electron acceptor character of the FR, and by the electron donor character of the antioxidant. In fact the relationship between them for any given pair FR-antioxidant has been rationalized in terms of the ionization energy (IE) of the donor and the electron affinity (EA) of the acceptor. Thus, it has been proposed that a necessary condition for the SET reactions to be viable is that IEdonor < EAacceptor. Based on this condition, a map known as the full electron donator acceptor map (FEDAM) was proposed (Fig. 2) that allows a quick and qualitative analysis of the possible electron flow in SET reactions.[41] Species at the lower left quadrant can be considered poor electron acceptors and good electron donors, while those at the upper right are poor electron donors and good electron acceptors. Accordingly, the electron flow is expected to occur from species located at the lower left to species located at the upper right of the map, which allows predicting which molecule is the most likely electron donor and electron acceptor in any considered pair. Therefore, based on their location in the FEDAM it is possible to predict which species would be good free radical scavengers, via SET.
It is important to note that even though the most common way in which the SET scavenging processes take place is with the electron being transferred from the antioxidant to the FR:
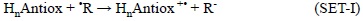
there are also cases when this process can occur in the opposite direction:
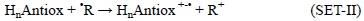
The relative position of the HnAntiox and 'R species in the FEDAM would allow anticipating the direction of the electron transfer. For example halogenated peroxyl radicals have relatively high IE and EA, thus they usually act as electron acceptors, i.e., they are scavenged by antioxidants via SET-I. In addition, the electro-accepting character of these radicals increases with the halogenation degree, and as a result the viability of the SET-I processes also increases with this feature.[254] SET-I pathways have been proposed as key routes for the free radical scavenging activity of the enol isomer of curcumin,[303] and highly galloylated tannin fractions.[304] In addition it is believed to be particularly important for the reactions of edaravone derivatives with some radicals such as 'OH, 'OCCl3 and CH3COO',[305] planar catechin analogues with peroxyl radicals,[306] resveratrol with oxygen radical,[307] and carotenoids with CCl3OO' [308, 309] and 'NO2 [291, 310]. The SET-II pathway, on the other hand, has been proposed to be involved in the reactions of the superoxide radical anion (O2'-) with carotenes[311] and xanthones,[312] and in the reactions of the NO radical with uric acid, caffeic acid, trolox and genistein. [313]
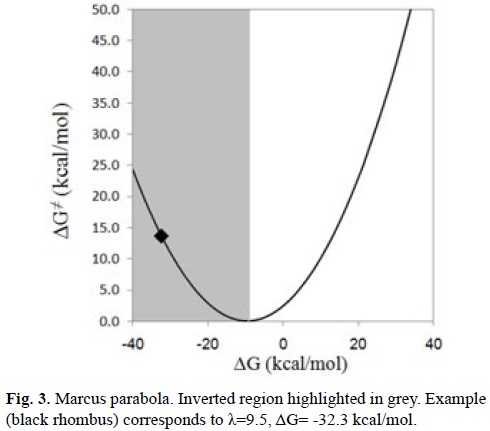
An important aspect of the SET processes that cannot be analyzed based only on IE and EA considerations is that when these reactions are highly exergonic, they can be located in the inverted region of the Marcus parabola (Fig. 3). Within this region the reaction barriers increase as the Gibbs energies of reaction (ΔG) become more negative. In other words, large negative ΔG values may correspond to rather slow processes. This behavior arises when ΔG is much lower than minus the reorganization energy (ΔG << -λ) yielding relatively high reaction barriers. Consequently, to take this into account is necessary to investigate the SET reactions using kinetics.
In addition SET as an isolated reaction pathway, responsible for antioxidant activity, is seldom found. It is much more common to find this kind of reaction taking place in conjunction with some other chemical processes. More details on this point are provided in section 4.2.
4.1.3. Hydrogen Atom Transfer (HAT)
This reaction mechanism corresponds to the transfer of a hydrogen atom, in a single step, from the antioxidant to the free radical:
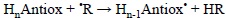
At this point it seems important to emphasize that is not trivial to differentiate between HAT and proton coupled electron transfer (PCET), so it is possible that a reaction assumed to take place via HAT can actually occur via PCET. More details on their differences, and the strategies to properly distinguish between these two mechanisms, are provided in the next section.
HAT has been reported to play a crucial role in the antioxidant activity of a large amount of chemical compounds. Its role is particularly important for phenolic compounds in their neutral forms, i.e., non-deprotonated. Therefore, the relative importance of the HAT mechanism is influenced by the environment. For example, it is usually the main reaction mechanism for the antioxidant activity of phenolic compounds in non-polar, lipid, environments where deprotonation processes are expected to be negligible, since such media do not provide enough solvation for the ionic species yielded by this process. In aqueous solution, the pH is the key factor determining the relative importance of the HAT mechanism for the antioxidant activity of phenols. If the pH is lower than the pKa of the phenolic compound it will remain mostly in its neutral form, thus increasing the importance of HAT. On the contrary, if the pH is higher than the pKa, deprotonation will occur, and the anionic species would be the preponderant one, thus decreasing the importance of HAT compared to any mechanism involving electron transfer from the phenolate ion, such as SPLET.
There are numerous studies supporting the essential role of HAT for the antioxidant activity of chemical compounds. In particular, the free radical scavenging activity of phenols, via HAT, has been well documented by both experimental and theoretical techniques. It has been proposed as a key reaction mechanism for polyphenols in general,[314] as well as for specific compounds such as procyanidins,[315] Maclurin,[316] 2,4,5-trimethoxy chalcones,[317] orientin,[318] cynarine,[319] silybin,[319] chlorogenic acid,[319] capsaicin,[320] α-mangostin,[321] fisetin,[322] baicalein,[322] the keto isomer of curcumin,[303] ellagic acid and its derivatives,[323] and some hydroxychalcones.[324] There are also some examples concerning the importance of HAT for the antioxidant effects of non-phenolic compounds such as lipoic and dihydrolipoic acids,[325] tryptophan and its derivatives,[326] glutathione,[327] and N-acetylcystein amide.[328]
Regarding the influence of the polarity of the environment, in non-polar media HAT has been identified as the principal reaction mechanisms, while other pathways become the most important ones in polar solvents for several compounds including alizarin and alizarin red S,[329] deoxybenzoins,[330] esculetin,[331] hydroxybenzoic[332] and dihydroxybenzoic[333] acids, fraxetin,[334] genistein,[335] daidzein,[335] glycitein,[335] equol,[335] 6-hydroxidaidzein,[335] 8-hydroxiglycitein,[335] resveratrol,[336] piceatannol[337] and other stilbenes,[338] hydroxychalcone,[339] morin,[340] quercetin and epicatechin.[341]
As mentioned before, pH also plays a role on the relative importance of HAT in the antioxidant activity of chemical compounds. To illustrate this point a particular example is used, the reaction of 'OOH with the protocatechuic acid (H3Prc). This acid has 3 pKa values (4.38, 8.74, and 10.67 [342]), which means that its dominant acid base form depends on the pH of the environment. In addition while HAT is the main mechanism of reaction for H3Prc and H2Prc-, SET becomes the most important pathway for HPrc2- and Prc3-. Accordingly the main mechanism for the overall 'OOH scavenging activity of proto-catechuic acid would also be influenced by the pH. HAT is the major scavenging mechanism at pH values < 4, while SET becomes the pathway contributing the most to the overall activity at pH > 6, and at 4 < pH < 6 both mechanisms are important (Fig. 4). This behavior is pretty common for phenolic compounds since as the phenolic moieties start deprotonating the most viable HAT reaction paths are no longer possible, and at the same time the formed phenolate ions are particular prompt to react via SET-I.
4.1.4. Proton Coupled Eleetron Transfer (PCET)
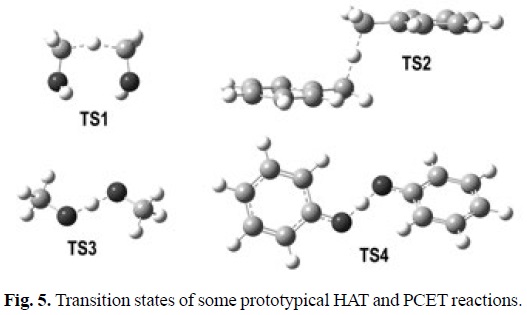
Since the PCET reactions yield exactly the same products as HAT, distinguishing between HAT and PCET is a non-trivial task. In HAT reactions the proton and the electron are transferred together as a single entity, i.e., a hydrogen atom. In PCET the electron and proton are concertedly transferred in a single step, without any stable intermediate, but as two separated particles. The main difference between these mechanisms is that while in HAT the donor and the acceptor are the same for the electron and the proton, in PCET they are different. That is why a commonly accepted way of describing PCET is a reaction involving a proton and electron transferred from different sets of orbitals. Therefore theoretical chemistry is a crucial tool to properly identify a chemical reaction as PCET, distinguishing it from HAT. Several strategies have been proposed for that purpose. Probably the most commonly used consists on analyzing the single occupied molecular orbital (SOMO) density surfaces of the transition states. For HAT reactions, they are expected to have significant density in atomic orbitals oriented along, or almost along, the donor-H-acceptor (transition) vector. In contrast, the SOMO of PCET transition states would involve p orbitals orthogonal to the transition vector, thus the proton is transferred between o orbitals while the electron is transferred between π orbitals.[344] In addition, albeit the presence of unshared electron pairs in the donor and the acceptor seems to be a requirement for PCET, such a presence does not assure that a PCET mechanism would prevail over HAT.
To illustrate the characteristics of PCET transition states, and compared them with those of HAT transition states, four chemical reactions are used here, which can be considered as prototypical examples:
1) Methanol + hydroxymethyl radical
2) Methanol + methoxl radical
3) Toluene + benzyl radical
4) Phenol + phenoxyl radical
The geometries of the transition states, and their SOMO density surfaces, are shown in Figures 5 and 6, respectively. The characteristic shape of the SOMO density surfaces for HAT and PCET transition states can be clearly appreciated in Fig. 6. For both HAT transition states the SOMO has significant density in orbitals lying on the donor-H-acceptor vector, and presents a node at the migrating H. On the other hand, for both PCET transition states there is no SOMO density on the donor-H-acceptor vector, i.e., there is a node on this vector, and the orbitals on the H donor and acceptor atoms are orthogonal to the transition vector. Using these distinctive features is then possible to identify if a reaction is actually HAT or PCET. It should be noted, however, that in some cases looking into orbitals deeper than SOMO may be necessary to identify the PCET mechanism. One example is the self-exchange reaction of the iminoxyl/oxime.[345]
Electronically adiabatic and nonadiabatic proton transfer processes can also be used to differentiate between HAT and PCET mechanisms, respectively.[346] Some quantitative diagnostics have been proposed to evaluate the degree of electron-proton nonadiabaticity, mainly based on following specific properties along the H coordinate. They are based on plots of the electronically diabatic and adiabatic potential curves, the component of the first-order nonadiabatic coupling vector between the first exited adiabatic electronic and the ground states along the H donor acceptor axis, the component of the dipole moment vector along the H donor acceptor axis, and the partial charges (obtained from the atomic charges derived from the electrostatic potential of the ground adiabatic state) of the transferring H, the acceptor molecule, and the donor molecule.[346] Using the topographical characteristics of the potential energy surfaces has also been demonstrated to be a successful strategy to differentiate between HAT and PCET. [347]
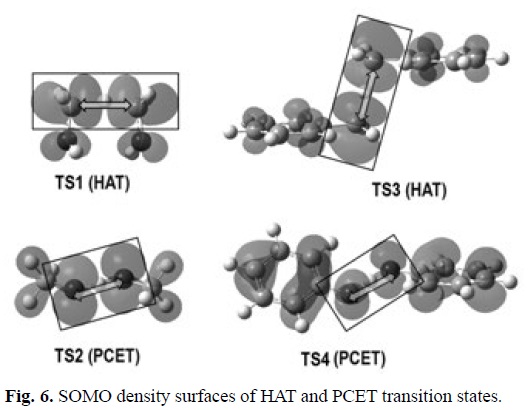
Following the idea of the charge descriptor, a simpler diagnostic is proposed here based on the analysis of the atomic charges of the H-donor, H-acceptor, and transferring H atoms, as a function of the reaction coordinate (s). The data was obtained using the points on the ground state reaction path (generated from intrinsic reaction coordinate, IRC, calculations) and the Hirshfeld partition scheme. In the diagnostic presented in reference [346] the charges on the acceptor and the donor molecules switch signs during the PCET reaction but not for HAT. In the case of the simplified descriptor what is important is not the sign change but only the shape of the curve. For HAT reactions the curve is very smooth along the whole reaction path, while for PCET the curve shows an abrupt jump around s=0.
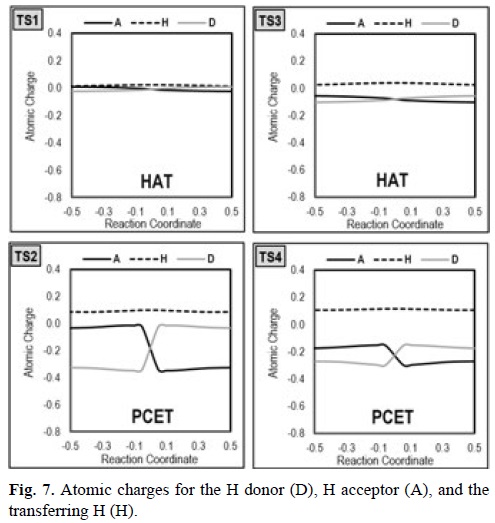
What is similar for both diagnostics is that for PCET the charge changes during the reaction are greater, and that the charge on the transferring H is more positive, compared to HAT.
A similar analysis may be performed using atomic spin densities, instead of atomic charges (Fig. 8). In this case the distinctive characteristic is also the curve shape, being smooth along the whole reaction path for HAT reactions, and presenting an abrupt jump around s=0 for PCET. In addition the spin density on the transferring hydrogen is higher for HAT than for PCET.
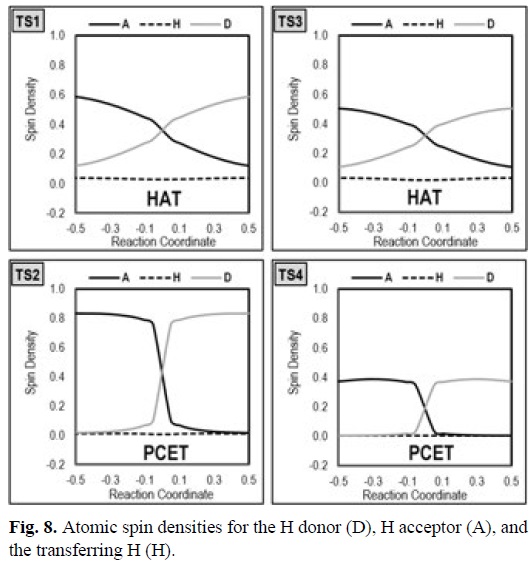
At this point it seems important to call attention to the fact that while in the prototypical systems presented here the distinction between HAT and PCET is clear with any of the above mentioned diagnostics, for other systems such a distinction may be not so evident. Thus it is always advisable to perform more than one diagnostic to assure that the reaction mechanism has been properly identified.
Regardless of the strategy used to identify PCET reactions, what is unquestionable is the importance that they have for chemical and biological processes. There are numerous cases where the PCET mechanism has been reported to be particularly relevant such as the H exchange in the tyrosyl/tyrosine couple, which is implicated in ribonucleotide reductase chemistry. [348] Some examples, regarding antioxidants, are the free radical scavenging activity of flavonoids,[349] the cardiovascular drug Dipyridamole,[350] and the quinone-hydroquinone system.[351] The PCET mechanisms also seems to play a crucial role in the antioxidant protection exerted by vitamin E and ubiquinol,[352] eupatilin,[353] diarylamines,[354] sulfenic ac-ids,[355-357] and halooximes of lawsone.[358]
4.2. Multiple Step Mechanisms
4.2.1. Sequential Proton Loss Eleetron Transfer (SPLET)
The SPLET mechanism was first proposed by Litwinienko and Ingold for the reactions between substituted phenols and the DPPH radical.[359-362] It consists of two steps, the first one corresponding to the antioxidant deprotonation, and the second one to a SET reaction, with the electron transferred from the deprotonated antioxidant to the free radical:
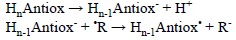
Even though it has been demonstrated that this mechanism is particularly important for the antioxidant activity of phenolic compounds,[363] it can also contribute to the protective effects of other compounds. This would depend mostly on two chemical characteristics of the antioxidant. The first one is its pKa, which would determine the proportion of the deprotonated species in aqueous solution, at each particular pH, for example at pH=7.4 under physiological conditions. The second one is the electron donating ability of the deprotonated antioxidant, and also the electron accepting ability of the free radical to scavenge. It is important to note that for SPLET to be the mechanism contributing the most to a particular antioxidant-free radical reaction it is not necessary that Hn-1Antiox- occurs to a larger extent than HnAntiox. Instead the condition that must be fulfilled is:
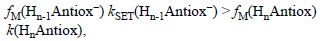
where fM(Hn-1Antiox-) and fM(HnAntiox) are the molar fractions of the deprotonated and non-deprotonated forms of the antioxidant at the pH of interest, kSET(Hn-1Antiox-) is the rate constant of the second step in the SPLET mechanism, i.e., of the electron transfer reaction from Hn-1Antiox- to the free radical, and k(HnAntiox) is the rate constant for the reaction between the non-deprotonated antioxidant and the free radical, regardless of the reaction mechanism involved.
In the particular case of phenolic compounds, this condition is usually satisfied when phenolate ions are yielded in the first step of the SPLET mechanism. This is because these ions are very good electron donors, which leads to very fast electron transfer reactions with various free radicals. However, not always the first deprotonation of a phenolic compound yield the corresponding phenolate ion. For example hydroxybenzoic acids present more than one acid-base sites: the carboxyl group, which deprotonates first; and the phenolic OH which is involved in the second pKa. Therefore, the carboxylate anions are the species formed after the first depronation, and the electro-donating ability of these anions is not high enough to promote fast electron transfer reactions towards most of the free radicals found in biological systems. In such cases the SdPLET (sequential double proton loss electron transfer) mechanism -which is just a particular case of SPLET- becomes the relevant process, since the second deprotonation, i.e., that yielding the phenolate ion, is the key to successfully complete the scavenging reaction.
It seems important to mention the role of the environment in the feasibility of SPLET pathways. First the solvent should be polar, and protic, to promote enough solvation for the deprotonated antioxidant to be formed. Therefore in biological systems this mechanism is expected to be important in the aqueous phase rather than in the lipid phase. As mentioned above, the second aspect of the surroundings that affect SPLET based mechanisms is the pH. As it increases so does the molar fraction of Hn-1Antiox-, and this increase in abundance is expected to promote the contributions of SPLET routes to the antioxidant activity of chemical compounds.
Nowadays, there is an overwhelming, and still increasing, amount of evidence supporting the key role of this mechanism on the protection against oxidative damage. SPLET has being identified as a crucial mechanism in the scavenging activity exerted by numerous compounds in polar environments. Some examples are curcumin,[360, 364] alizarin and alizarin red S,[329] deoxybenzoins,[330] esculetin,[331] hydroxybenzoic and dihydroxybenzoic acids,[332, 333, 365, 366] fraxetin,[334] genistein, daidzein, glycitein, equol, 6-hydroxidaidzein, and 8-hydroxiglycitein,[335] resveratrol,[336, 367] piceatannol,[337] and other stilbenes,[338] hydroxychalcones,[339, 368, 369] morin,[340] xanthones,[312] edaravone and its derivatives,[305] flavonoids,[370] vitamin E,[371] quercetin and epicatechin,[341] procyanidins,[315] kaemp-ferol,[372]Dp, 2,4,5-trimethoxy chalcones,[317] indolin-2-one derivatives,[373] Daidzein derivatives,[374] gallic acid,[375] erodiol,[376] silybin and 2,3-dehydrosilybin,[377] aminothiazol hydroxyl coumarin derivatives,[378] tocopheramines and tocotrienamines,[379] isoflavonoids,[380, 381] Trolox ,[382] stobadine derivatives,[383] 4-mercaptostilbenes,[384] chroman derivatives,[385-387] phenylpropanoid glycoside ana-logs,[388] a-pyridoin and its derivatives,[389] baicalein,[390] and purpurin.[391]
4.2.2. Sequential Eleetron Proton Transfer (SEPT)
This mechanism is also known as single electron transfer-proton transfer (SET-PT). It comprises an electron transfer reaction from the molecule to the free radical yielding the oxidized molecule, followed by the deprotonation of the later:
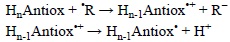
When the antioxidant is a neutral molecule, a radical cation is formed as the intermediate of this reaction. Therefore polar solvents are necessary for this mechanism to be important since they would promote the required solvation for the ionic intermediates yielded in the first step. In addition the electron donor ability of the antioxidants should be particularly high for such intermediates to be formed rapidly enough, so the overall reaction does not become too slow. It is also important to note that the environment is expected to strongly influence the viability of SEPT processes. Solvents not only need to be polar but also protic due to the nature of the second step of this mechanism. pH is also important since it rules deprotonation, i.e., the more basic the pH the higher the viability of the second step. In addition, the possible presence of strong bases in the surroundings would have similar effects, because of their ability of acting as H+ acceptors.
There are some reports on the role of SEPT in the antioxidant activity of chemical compounds, albeit they are significantly less abundant than those focused on HAT, PCET and SPLET mechanism. For example SEPT has been reported to be important for the antioxidant ability of baicalein,[392] astaxanthin and its n-octanoic monoester and diester,[393] and for quercetin, provided that it is in the presence of bases that have HOMO energies lower than that of the SOMO of the quercetin radical cation.[394] It has also been identified as the main route in the DPPH and galvinoxyl radical scavenging activity of vitamin E models,[395] and in theroxyl radical-scavenging process of a-tocopherol.[396]
However, SEPT is not only involved in the antioxidant activity of chemical compounds but also on the oxidative damage inflicted to biomolecules by reactive radicals such as 'OH. For example it has been demonstrated, in a theoretical study, that SEPT is the main reaction channel involved in the guanosine + 'OH reaction,[243] which allowed to explain the associated UV-Vis experimental data. SEPT was also identified as the mechanism responsible for the oxidation of 2'-deoxyguanosine sites in double-stranded DNA,[171] and for the reaction of triplet excited state of ketoprofen derivatives with amino acids and nucleosides.[397]
4.2.3. Sequential proton loss hydrogen atom transfer (SPLHAT)
This mechanism consists of two steps, the first one is identical to that of the SPLET process and yields the deprotonated anti-oxidant, while the second one differ from SPLET in the particle that is transferred which is an electron in SPLET and an H atom in SPLHAT:
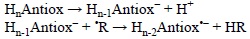
This mechanism has been mentioned in the literature only once, explicitly using the SPLHAT name,[398] for anthocyanidins. However, its importance in the free radical scavenging activity for other compounds has been also described. For example this is the main mechanism in the reactions of esculetin with 'OOCH3 and a model of lipid peroxyl ('OOCHCH2) radicals,[331] and for the reaction of gallic acid with 'OH.[399] It has also been reported to be significant for the free radical scavenging activities of α-mangostin,[321] ellagic acid,[254] propyl gallate,[400] caffeic and other phenolic acids.[401]
SPLHAT is expected to compete with the SPLET mechanism, since they have the first step in common. Therefore any environmental factor contributing to increase deprotonation would favor both processes. Therefore their relative importance would be ruled by the viability and rate of the second step. This means that it would depend on the facility of the deprotonated antioxidant for transferring an H atom or an electron. The higher the electron donor ability of the deprotonated antioxidant the higher the probability of SPLET to be more important than SPLHAT. On the contrary, those species with more labile H atoms would favor SPLHAT over SPLET. The relative importance of these two routes are also expected to be influenced by the chemical nature of the reacting free radical. As the electron acceptor capability of the free radical increases, so does the relative importance of SPLET.
5. Computational Strategies
There are numerous computational strategies that can be used to study the antioxidant activity of chemical compounds. Here they are grouped into three large categories depending on the kind of calculated data. Intrinsic reactivity based strategies deal with only one species, the antioxidant. Within this strategy, molecules with potential antioxidant activity are analyzed by quantifying some properties that characterize their intrinsic reactivity. To that purpose specific chemical processes are chosen in such a way that the calculated properties can be associated with a particular mechanism of reaction. This way numerical comparisons can be performed and used to suggest which molecule, or molecules, are expect to exhibit the best antioxidant capacity. Thermochemical based strategies consist of calculating the energies, usually enthalpies (albeit Gibbs energies would probably be a better choice), of particular chemical reactions involved in the different reaction mechanisms associated with the free radical scavenging processes. That way not only the reactivity of the antioxidant is taken into account, but also that of the reacting free radical. These two categories constitute the most abundant kind of theoretical studies aiming to propose antioxidant trends.[317-319, 323, 324, 326, 329, 332, 349, 380, 381, 402-418] The third category corresponds to calculations of kinetic data, especially rate constants, that can be directly compared with experimental measurements.
5.1. Intrinsic Reactivity Based Strategies
5.1.1. Bond Dissociation Energies (BDE)
They are usually calculated for the dissociation of bonds involving hydrogen atoms, and are associated with the predisposition of a compound to react via HAT. The most common way of reporting BDEs is using the hypothetical reaction:
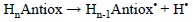
and calculate the corresponding electronic energy, including or not zero point (ZPE) corrections, as:
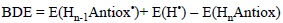
Then comparing the BDE values for a set of molecules it can be predicted which one should be more reactive via HAT, i.e., the lower the BDE the more reactive the compound.
The BDE term has also being used in the literature for referring to bond dissociation enthalpies. This approach is almost identical, the only difference is that in this case temperature effects (to enthalpy) are included and the energy difference is obtained as:
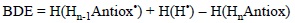
5.1.2. Ionization Energies (IE)
They are usually calculated for the first ionization process and are associated with the propensity of a compound to react via SET. IE values can be calculated using different approaches, with the most frequently used corresponding to vertical energies. The simplest of these strategies is based on the Koop-mans-theorem[419] or the Perdew-Levy[420] approximations for Hartree-Fock (HF) and density functional theory (DFT) based methods, respectively. Within this approach the IE can be obtained as:
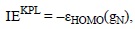
where ξHOMO(gN) is the energy of the highest occupied molecular orbital (HOMO) of the N-electron system (HnAntiox), at its optimized geometry (gN). Within this approach only one calculation is required, that of the molecule of interest.
Another strategy, referred to as AE, or the indirect, approach, can also be used. In this case the IE values are obtained from the following expression:
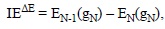
where EN(gN) is the total energies of the N-electron system and EN-1(gN) is the energy of the (N-1) electron system (HnAntiox'+), both calculated at the gN geometry. This strategy implies a second calculation, the total energy of the ionized species (with N-1 electrons) at the geometry of the N-electron parent molecule.
IE values can also be estimated using methods based on the electron propagator theory (EPT).[421, 422] They are reliable and efficient tools allowing direct estimation of vertical ionization energies from a single calculation that usually are more accurate than the above mentioned ones.
Adiabatic ionization energies can also be obtained by including the geometry relaxation of the N-1 species, according to:
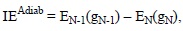
Therefore this strategy requires optimizing not only the geometry of the molecule of interest, but also that of its oxidized form.
Regardless of the strategy used to obtain the IE values, trends can be established for the donating ability of a set of compounds. Thus, based on these values, the species more prompt to react via SET can be identified. It is important to note, however, that for such comparisons to be successful the IE values of all the compared molecules must be obtained using the same approach. In addition, the predictions made this way should be taken with caution, since (as mentioned in section 4.1.2) IE alone can be misleading if the SET process lies on the inverted region of the Marcus parabola. In addition, while IE are defined in gas phase, to interpret them in the context of oxidant/antioxidant activities the proper solvent should be include. An even better approach, more easily comparable with experimental data, would be to estimate redox potentials.
5.1.3. Proton Affinities (PA)
Since PA values are directly related to the tendency of a molecule to deprotonate, they can be used to identify, from a set of molecules, those that are most likely to be involved in the first step of the SPLET and SPLHAT mechanisms. They are usually obtained as the reaction energy of:
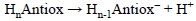
PA is defined as the negative of the enthalpy change in a, real or hypothetical, gas phase reaction between an electrically neutral chemical species and a proton to give the conjugate acid of the former. Therefore the most appropriate way to calculate this property is using enthalpy values:
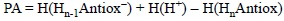
The lower the PA the most likely the deprotonation of HnAntiox. Even though proton affinities are defined in gas phase, when calculating this quantity for assessing antioxidant activity a modification can be introduced by including protic polar solvents in the modeling, so the results are more in line with the task at hand. The same applies for any other calculation of chemical process involving charged species.
5.1.4. Proton Dissoeiation Enthalpies (PDE)
This quantity has been specifically designed in connection with the second step of the SEPT mechanism. The only difference between PDE and PA is that the latter is defined for the neutral form of the molecule of interest, while PDE is defined for the deprotonation process of the radical cation yield by the first step of SEPT:
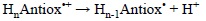
Accordingly, PDE can be obtained from the following expression:
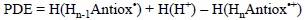
In line with the analysis of PA values, the lower the PDE the most likely the deprotonation of HnAntiox'+, i.e. the most likely the second step of the SEPT process.
5.2. Thermochemical Based Strategies
5.2.1. Eleetron Transfer Enthalpies (ETE)
ETE values correspond to the enthalpies of SET reactions between any given pair of antioxidant and free radical. Thus they are calculated as:
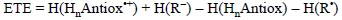
This quantity differs from IE in the explicit inclusion of the reacting free radicals. Therefore it offers a more complete picture involving not only the tendency of a particular antioxidant to donate one electron, but also the ability of the free radical to accept it. In addition, while IE is defined for gas phase, the ETE values are usually obtained including solvent effects, so the chemistry involved is closer to the actual free radical scavenging activity that antioxidants may present in biological systems. ETE has another advantage over IE, it has a meaningful sign that can be directly related to the viability of the reaction of interest. As it is the case for any other chemical reaction, if ETE values are negative the process is exothermic and if they are positive the process is endothermic. Since entropic changes in SET reactions are expected to be negligible, then negative ETE are necessary for the reaction to take place. Moreover, the more negative the ETE, the more thermochemicaly feasible the reaction. Accordingly, by using the same free radical it is possible to establish trends in reactivity for a series of potential antioxidants. In the same way, calculating ETE values for a particular molecule and a set of free radicals, makes possible to propose which of them could be better scavenged by the molecule of interest. Therefore two kinds of trends can be obtained from ETE analyses.
5.2.2. Proton Transfer Enthalpies (PTE)
PTE values can be used to include the possible influence of any base present in the biological media that may promote deprotonation, thus favoring the first step of the SPLET and SPLHAT processes. Therefore while PA and PDE values are useful to asses trends in deprotonation to the environment, i.e., mainly the solvent, PTE allows including the potential effects of other species present during the free radical scavenging process. The chemical reaction associated with PTE would be:
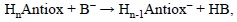
where B- represents the base, and HB its conjugated acid. The PTE value is then obtained as:
PTE = H(HnAntiox-) + H(HB) - H(HnAntiox) - H(B-)
As it is the case for ETE, the sign of the PTE value can be taken as a criteria of the feasibility of the proton transfer, and, logically, the stronger the base the more likely the reaction. Enthalpy is a good enough criteria in this case since no significant entropy changes are expected for reactions with identical molecularity for reactants and products. Including solvent effects in this kind of calculation is recommended. PTE is the only index that allows including the influence of species other than those directly involved in the free radical scavenging process on the viability of such process.
5.2.3. Hydrogen Transfer Enthalpies (HTE)
This quantity corresponds to the enthalpy of a HAT reaction for any antioxidant - free radical pair of interest. Therefore it can be calculated as:
HTE = H(Hn-1Antiox') + H(HR) - H(HnAntiox) - H(R')
The main difference between HTE and BDE is that the influence of the chemical nature of the reacting radical is explicitly included that in HTE, i.e., HTE takes into consideration not only the H donating ability of the antioxidant, but also the H accepting ability of the free radical. Similarly to what happens in the reactions used to calculate ETE and PTE, in this case entropy changes are expected to be only minor. Thus enthalpy changes are enough to assess thermochemical viability, which is the first criterion to predict if a HAT reaction would be possible. Direct comparisons among HTE values corresponding to the reactions of different molecules with the same radical are useful to identify reactivity trends. The more negative the HTE, the more viable the HAT reaction.
5.2.4. Other Properties
The thermochemical properties discussed above are, probably, the most frequently used regarding thermochemical approaches. However, many others can also be used to investigate the thermochemical feasibility of any particular reaction potentially involved in the antioxidant activity of chemical compounds. To that purpose the idea is to identify the reaction of interest and then performed the necessary calculation to estimate not only the corresponding enthalpies, but also the Gibbs free energies, or the ZPE corrected energies. For example this strategy may be applied to the RAF mechanism, or to the overall processes that comprises more than one elementary reaction step. It can also be helpful for modeling oxidative damage to molecules of biological interest, antioxidant regeneration processes, or repairing of damaged targets. It allows including the effects of the solvent polarity or the catalytic effects of some component in the environment. When Gibbs free energies are used it also allows to account for entropic effects.
5.3. Kinetics Based Strategies
Both, intrinsic reactivity and thermochemical based strategies are very useful and usually provides first physicochemical insights of the different reaction mechanisms. Intrinsic reactivity strategies are especially advantageous for evaluating large numbers of similar antioxidants. This is because the associated computational protocols are not particularly expensive or laborious. Thermochemical strategies provide information on the influence in reactivity of every reactant involved, as well as of the solvent. However there are a number of aspects related to the quantification of the antioxidant capacity (AOC) that cannot be taken into account using them.
They may not be enough for analyzing AOC trends, for example, when the Bell-Evans-Polanyi principle is not ful-filled.1 Another example is the analysis of competing parallel reactions, such as HAT and RAF. In this case the entropy changes for both processes are different, usually they are small for HAT while they are significant for RAF. In addition tunneling effects can be important for HAT reactions, while they are expected to be negligible for RAF when the radical involved is other than 'H. Thus the entropy influence and the quantum effects would be crucial to identify the preponderant mechanism. AOC can also be significantly affected by the dynamic effects that become relevant in transition states and by the potential role of the pH on reactivity. For antioxidants with acid sites, their pKas would regulate the proportion of the different acid-base forms (cationic, neutral, mono-anionic, di-anionic, etc.) at each particular pH. Moreover, depending on the corresponding molar fractions, the main reaction mechanism and the overall AOC can be substantially affected. In addition for almost any possible antioxidant more than one reaction channel may contribute to the overall reactivity. Accordingly, the only way of performing analyses that are in line with past or future experimental evidences is to include them all simultaneously and appropriately weighting them. In addition, as mentioned before, for antioxidants to be able of efficiently protect biological targets they must react faster than the molecules that are aimed to protect. Thus kinetic approaches become particularly important in AOC studies, since they would account for the above described aspects, provided that they are properly carried out.
Probably the simplest way of including kinetics in theoretical studies is by calculating reaction barriers. However, this way there are some aspects that cannot be accounted for. Some examples are the possible tunneling effects, and the analyses of the overall reactivity by simultaneously considering the contributions of all the reaction channels. Therefore this review will be focuses in a particular computational protocol, specifically designed to be used as an AOC test, which is known as the quantum mechanics-based test for overall free radical scavenging activity (QM-ORSA).[343]
5.3.1. The QM-ORSA Protoeol
This protocol comprises several computational aspects, specially designed to facilitate comparisons among the calculated data, as well as to assure their accuracy. The key points of the QM-ORSA protocol are:
1. Always using the same eomputational methodology. The recommended electronic structure methods are those within the framework of the DFT because of their excellent balance between computational cost and accuracy. In particular the LC-ωPBE, M06-2X, BMK, B2PLYP, M05-2X, and MN12SX approaches, since they have been recently demonstrated to be the best performing functionals for kinetic calculations in solution.[423] Concerning the basis set, it is crucial to use at least a double Z, including polarization and diffuse functions for non-hydrogen atoms. In addition, diffuse functions becomes especially important when studying compounds with anionic species involved in their AOC. Therefore basis sets from 6-31+G(d) to 6-311++G(d,p) are expected to be good enough, and choosing one of them would mainly depend on the size of the system under study. Additionally, and at risk of sounding repetitive, it seems relevant to insist on the fact that gas phase (vacuum) calculations are not appropriate for modeling chemical reactions with the intention of being interpreted regarding OS or AOC. Solvent continuum models are probably the most suitable ones for that purpose, since they do not significantly increase computation times. The one used within the QM-ORSA protocol is the solvation model based on density (SMD), since it is considered universal, applicable to any uncharged or charged solute in any liquid medium or solvent.[424]. The recommended solvents are water and pentylethanoate to mimic aqueous and lipid environments, respectively, albeit other solvents can be chosen to mimic the hydrophobic phase.
2. Modeling all meehanisms and reaetion sites. Ussualy, a single antioxidant is able of reacting through more than one mechanism and at more than one reaction site. They all must be considered, since this is the only way of quantifying their possible contributions to the overall AOC of the species of interest. Depending on the antioxidant, this may become a very laborious task. One possible way to reduce it is to evaluate first the thermochemical viability of all the reaction pathways using their Gibbs free energies of reaction (ΔG). Then, using these data to identify the exergonic and isoergonic reaction paths (ΔG ≤ 0), and consider only them for kinetic calculations. This simplified strategy is based on the assumption that endergonic pathways would be reversible and therefore the formed products will not be observed. However, it is important to note that such pathways might still be important if their products rapidly react further. Thus, in such cases they still should be included in the kinetic study. SET reactions frequently show such a behavior. That is why it is recommended to consider them even when their ΔG values are positive, but low (≤ 10 kcal/mol).
3. Modeling reaetions with the same free radieal. This is a key aspect of the protocol since reactivity, and thus rate constants, are expected to be significantly influenced by the chemical nature of the reacting free radical. It is recommend to use free radicals of low to middle reactivity for studying the relative AOC of chemical compounds,[289, 425] such as the hydroperoxyl radical (HOO-'). This is because free radicals of high reactivity, like 'OH, can react with a wide variety of compounds at similar rates (close to the diffusion-limit). Thus, comparisons based on the reactions of such radicals might lead to miss-conclude that all the analyzed compounds have similar AOC, while that might not be the case when a wider spectrum of free radicals is considered. In addition, such highly reactive free radicals cannot be efficiently intercepted in biological systems, since they would immediately react with almost any molecule near their formation place, and with little selectivity toward the different reaction sites.
4. Using the transition state theory (TST) for ealeulating the rate eonstants of eaeh reaetion ehannel. The main benefit of using conventional TST is that it needs only a few information on the reaction potential energy surface (PES). i.e. calculations of stationary points (reactants and transition states) would be enough. This makes TST an appealing choice, particularly for chemical systems of relatively large size. Despite of its apparent simplicity, this is a reliable theory which has been shown to produce rate constants of radical-molecule reactions with uncertainties similar to those arising from experiments.[343] For particular cases that may need a more sophisticated methodology, the Interpolated Variational Transition-State Theory by Mapping (IVTST-M)[426] is recommended.
5. Using the Mareus Theory to estimate the reaetion barriers of SET reaetions. Since transition states for SET reactions are not driven by nucleus motion, they cannot be located using electronic structure methods. Therefore a different strategy is necessary to obtain the reaction barriers for the corresponding TST calculations. The Marcus theory[427, 428] can be used for that purpose. It allows calculating the barrier of any SET reaction from two thermodynamic parameters, the nuclear reorganization energy (λ) and the free energy of reaction.
6. Taking into aeeount the reaetion path degeneraey (σ). It is also known as the statistical factor that accounts for the number of equivalent reaction paths, i.e., the different but equivalent ways in which a reaction may occur. It can be estimated by considering all identical atoms and counting the number of different, but equivalent, ways in which they can be arranged by rotation. Another way to estimate σ is by the strategy proposed by Pollak and Pechukas,[429] which is based on using the total symmetry numbers of the transition state and the reactants. Even though this approach is valid in most cases, there are some exceptions. The interested reader is referred to the work by Fernandez-Ramos et al.[430] for further information on this subject, including examples. When estimating σ, attention should be paid to avoid double-counting. Thus, if symmetry constrains were imposed in the calculations of transition states or reactants, this must be considered in the calculations of σ.
7. Ineluding tunneling eorreetions. There are numerous reactions, which involve light particles that may present significant quantum effects. Thus ignoring them would lead to rather large errors in the rate constants, calculated with TST. For example not including tunneling corrections might cause substantial underestimations of rate constants for HAT reactions with barriers of moderate height. Consequently, a posteriori corrections are frequently necessary to amend this omission. Tunneling corrections are probably the most frequently used, since they accounts for the main quantum effects in chemical reactions. i.e. penetration through the barrier. A more complete inclusion of quantum effects can be achieved by calculating transmission coefficients, which also corrects for the non-separability of the reaction coordinates and for non-equilibrium reactants. A detailed analysis on this point is not within the scope of this review, but it can be found elsewhere.[431] To compute tunneling corrections the one-dimensional zero-point-inclusive Eckart surfaces can be used in combination with TST calculations, provided the calculations are at room or higher temperatures, since they both require just the same information on the PES.[432, 433] For the same reason, the small-curvature tunneling (SCT)[434] is recommended when rate constants are calculated using IVTST-M, or other variational TST method.
8. Using 1 M standard state. The results produced by most of the currently available computational codes correspond to the 1 atm standard state, i.e. to the gas phase. On the other hand, the calculated rate constants are mostly for bimolecular reactions, in solution, thus their expected units are M-1s-1. Therefore the appropriate conversion should be made to assure that the calculated values are directly comparable with the experimental ones. This is particularly important when the calculated energies involve processes with changes in molecularity, such as RAF reaction energies, and especially the energy barriers (ΔG*) for reactions with more than one reactant. For example, for bimolecular reactions, at 298K, such a conversion decreases ΔG* values by 1.89 kcal/mol, which implies that not using the 1 M standard state would lead to artificial underestimations of the rate constants by about 24.3 times.
9. Correeting for diffusion-eontrolled rates. Calculated rate constants (k) can be close to the diffusion-limited regime for chemical reactions involving very reactive species. Even though it might seem obvious, it is important to insist on the fact that diffusing within the solvent would limit the rate at which any encounter between reactants can occur in solution. Accordingly, rate constants higher than the rate of diffusion would lack physical meaning. Furthermore, calculated rate constants intended to predict or reproduce the actual behavior of real systems, must be directly comparable with those observed under experimental conditions. Therefore, in cases within -or close to- the diffusion-limited regime they cannot be directly obtained from TST calculations, since the role of diffusion is crucial and must be taken into account. To that purpose the Collins-Kimball theory[435] can be used, in conjunction with the steady-state Smoluchowski[436] and the Stokes-Einstein[437, 438] approaches.
10. Caleulating total rate eoeffieients and branehing ratios. The total rate coefficients (ktot) for each chemical entity in any solvent, for example the neutral and an-ionic forms of the antioxidant in aqueous solution, are calculated as the sum of the apparent rate constants of all the corresponding reaction paths. After calculating the total rate coefficients this way, the relative amount of products (%) yielded by each reaction path can be easily estimated using branching ratios. This information constitutes one of the strengths of theoretical calculations, because it is frequently very difficult to obtain from experiments, and allows quantifying the importance of each individual reaction path, and thus the proportion in which each product is formed.
11. Caleulating overall rate eoeffieients. The overall rate coefficient (koverall) for chemical reactions in non-polar media is frequently equal to ktot. Exceptions may appear when there is more than one nuclear configuration (for example tautomers) with similar energy, i.e., at significant concentrations. On the other hand, in aqueous solution, it is common to find more than one acid-base form of free radical scavengers, with non-negligible contribution to the overall AOC, at physiological pH. This, logically, would depend on their pKa values, which rules the population of each acid-base form. These populations can be quantified by calculating the corresponding molar fractions (Mf), which are used to estimate koverall. To that purpose the Mf.ktot product is calculated for each relevant acid-base species, and then koverall is obtained by summing up all of them.
12. Using a threshold value to identify eompounds with signifieant primary AOC. Once the overall rate coefficient is obtained for a particular chemical compound it is necessary to decide what this value means in terms of antioxidant protection. The first aspect to analyze should be whether the studied compound can be considered as a primary antioxidant. To that purpose the key point to evaluate if it would react faster, with a particular free radical, than the biological targets to protect. Within the QM-ORSA protocol, the recommended radical for evaluating AOC is OOH. The reasons of that choice are provided in section 6.2. The rate constants corresponding to the damage caused by this radical to polyunsaturated fatty acids has been measured to be 1.18-3.05x103 M-1s-1.[439] These values do not include the molar fraction of HOO, since they were measured at acid pH where Mf(HOO) is expected to be equal to, or very close to, one. The 1.18x103 M-1s-1 is used as the threshold value (kthresh) for assessing the efficiency of compounds as OS protectors. Compounds with koverall < kthresh are not considered as efficient primary antioxidants; while compounds with koverall ≥ kthresh are proposed as effective to that purpose. In this analysis polyunsaturated fatty acids are assumed as the biological targets to protect. Other biological targets, such as DNA and proteins, can also be used as reference. However, the reactivity of most of them towards free radicals is, fortunately, lower than that of bis-allylic hydrogens in polyun-saturated acids.[440] Therefore, any compound able of protecting polyunsaturated fatty acids from free radical damage, is also expected to protect other -less or similarly reactive- biological targets.
13. Making separated eomparisons for non-polar and polar environments. It is recommended to establish AOC trends separately considering hydrophilic and hydro-phobic environments. This is because the reactivity of chemical species can be significantly influenced by the environment. In addition, and for the same reason, when interested in reactions that take place in aqueous solution, comparisons should be performed based on rate constants obtained at the same pH.
14. Establishing trends. Trends in the AOC activity can be proposed using two different strategies when using the QM-ORSA protocol. The first one, referred to as the absolute criterion, consists of performing direct comparisons based on the overall rate constants. Logically, the larger the koverall value the higher the primary AOC of the analyzed compound. The second one, referred to as the relative criterion, uses a particular antioxidant as a reference, against which the AOC of other compounds is compared to. Since Trolox is probably the antioxidant most frequently used to that purpose in experimental assays, it seems logical to use it also within QM-ORSA protocol. Its overall rate coefficients in hydrophobic and hydrophilic environments have already been calculated using his protocol. [382] Thus, the relative primary AOC, of any antioxidant of interest, can be expressed as the ratio between its koverall and that of Trolox (under the same conditions, i.e., kind of solvent and pH in aqueous solution). This kind of comparison is also expected to maximize the cancelation of errors inherent to any calculation.
When the QM-ORSA protocol is properly followed, it is expected to provide reliable information on different aspects related to the primary AOC of chemical compounds. It would allow identifying the main mechanisms, and reaction sites, involved in the free radical scavenging activity of antioxidants, as well as quantifying their primary AOC in both polar (aqueous) and non-polar (lipid) media. It can be used to provide two different scales for quantification: (i) the absolute, based on overall rate coefficients, and (ii) the relative, using Trolox as reference. These kind of analyses are expected to facilitate direct comparisons with experimental data. Using this protocol it is possible to establish trends in the primary AOC activity for both hydrophilic and hydrophobic environments, which may help identifying the most efficient primary antioxidants, as well as their structural features. This information might, hopefully, help designing efficient pharmacological strategies against OS.
5.3.2. Validation by Comparison with Experimental Data
To assess the reliability of the QM-ORSA protocol, 25 rate constants calculated using it have been compared with the available experimental data (Table 1). The experimental pH has been included in the calculations by using the molar fractions of the different acid-base forms when calculating koverall. In this regard there is a free radical that deserves particular attention, the hydroperoxyl radical (HOO'). It has a pKa = 4.8, which means that its equilibrium with the corresponding conjugated base (O2'-) would be affected by the pH (Fig. 9). For example, while at pH=3 the molar fraction of HOO' is 0.9844, at pH = 7.4 it drastically lowers to 0.0025. Thus, to reproduce experimental rate constants of reactions involving this radical, this is a crucial aspect that should be included.
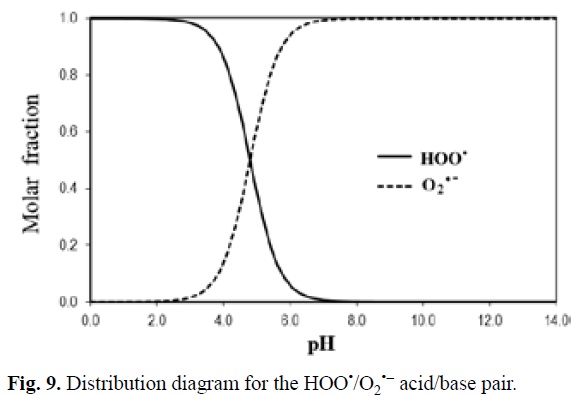
The reaction between ascorbic acid and the HOO'/O2'-pair is a good example to illustrate this point. The koverall calculated for this reaction is ~1.0x108 M-1 s-1, without considering the acid-base equilibrium of HOO. This value is significantly apart for the experimental measures. However after including the HOO' molar fraction at each pH of interest, the agreement between the calculated and the experimental values is very good at a wide range of pHs (Table 1).
The largest discrepancy between calculated and experimental rate coefficients was found for the adrenaline + HOO'/ O2'- reaction (keale 10.2 times lower than kexp). However in most cases (19 out of 25) the ratio between keale and kexp is lower than 3. In addition, it should be noted that discrepancies similar to the largest one found here, can also arise from experimental measurements. For example, for the glutathione + 'OH reaction the reported experimental values range from 3.48x109 to 4.4x1010 M-1s-1,[459-461] i.e., one experimental value is 12.6 times larger than the other.
In general the agreement found between the experimental rate coefficients and those obtained with QM-ORSA is excellent for the whole test set of reactions (Fig. 10). In fact, the correlation of log(keale) -obtained with the QM-ORSA protocol- vs. log(kexp) shows that not only the R2 value (0.97) is close to one, but that also the slope is close to one (0.98), and the intercept is close to zero (0.05). This correlation demonstrated the reliability of the presented methodology for providing rate coefficients that can be directly compared with the experimental values, and that can also be used to predict such values. Moreover, considering the compared data altogether, it can be stated that the reliability of the rate coefficients obtained using the QM-ORSA protocol can be comparable to that of experimental measurements.
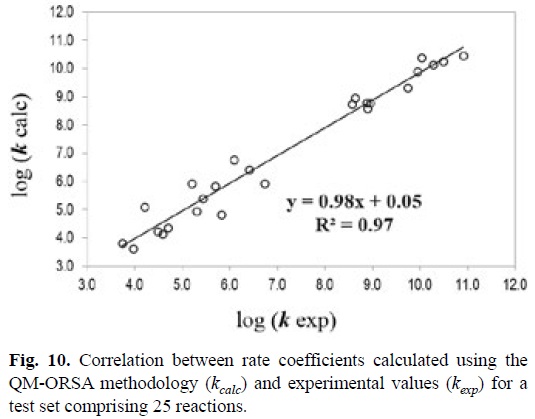
6. Trends in Activity
It is common knowledge that natural products are complex mixtures comprising diverse chemical compounds. Therefore, when natural products show good antioxidant activity it would be desirable to identify which of their components are mainly responsible for such activity. This is because under high OS conditions, the diet may not be enough for providing all the necessary antioxidants. Under such conditions dietary supplements would be a good choice, but for producing them wisely a necessary previous step is to know which compounds would exert the best protection against OS, so they are the ones included in the supplements.
6.1. Experimental Approaches
There are several experimental assays available that can be used to measure the antioxidant capacity (AOC) of natural products or specific chemical compounds. Some of the most used ones are the DPPH (2,2-diphenyl-1-picrylhydrazyl), the ferric reducing antioxidant power (FRAP), the Folin-Ciocalteu reagent (FCR), the oxygen radical absorbance capacity assay (ORAC), the total radical-trapping antioxidant parameter (TRAP), and the Trolox equivalent antioxidant capacity (TEAC) or other ABTS assays. Albeit detailed descriptions of these assays can be found elsewhere,[475-478] some of their features are presented in Table 2 to facilitate the further discussion.
Despite of the variety of experimental assays available to measure AOC, the current lack of a universal AOC assay has been pointed out by Prior et al.[475] This concern is shared by other authors, such as Frankel and Meyer, who have stated that "...there is no simple universal method by which AOC can be measured accurately and quantitatively", and that "There is a great need to standardise antioxidant testing to minimise the present chaos in the methodologies used to evaluate antioxidants".[476] The reasons for this can be rationalized based on the chemical reactions and experimental conditions used in these assays. For example, while HAT is the main reaction mechanism involved in ORAC and TRAP assays (Table 2), FRAP is essentially governed by SET, and so are TEAC and DPPH assays albeit it has been proposed that HAT reactions can also contribute to both of them.[475, 487] This means that a previous knowledge on the main reaction mechanism for a particular antioxidant is necessary before choosing the most appropriate assay. For example glutathione it is a poor electron donor that exerts its antioxidant protection mainly via HAT.[327] Consequently, while TRAP and ORAC would be suitable for evaluating its AOC activity, using the FRAP assay might lead to erroneously conclude that it is not efficient as antioxidant.
In addition, the chemical nature of the reacting free radical and the environment, may alter the reactivity of a particular compound towards free radicals, as well as the main mechanism of reaction. The environment can alter not only the trends in AOC but also the concentration of a particular antioxidant, depending on its solubility. It has been reported that in bulk oil systems polar antioxidants are more active, while in lipid/water emulsions non-polar antioxidants are more effective, which is known as the polar paradox.[488] Some particular examples, regarding the influence of the environment on AOC trends have been reported by Frankel et al.[489]. They found that the AOC trend changes from Trolox > α-tocopherol, ascorbic acid > ascorbyl palmitate, in bulk corn oil, to α-tocopherol > Trolox ~ ascorbyl palmitate > ascorbic acid in oil/water emulsion. In another work from the same group[490] it was found that the trend carnosic acid ~ rosmarinic acid > carnosol, in bulk corn oil, becomes carnosol > carnosic acid > rosmarinic acid, in oil/ water emulsion. In the same work it was reported that AOC can be also influenced by pH. For example, it decreases as the pH goes from 4 to 7 for carnosic acid and carnosol. It was also observed that, depending on the method of choice for measuring AOC (hexanal or hydroperoxide formation), the relative AOC of a-tocopherol and Trolox changes. In addition, due to the importance of phenolic compounds as antioxidants there are tests, such as FCR, that are based on measuring total phenolic content, which is then directly associated with AOC. However, not all phenolic compounds are equally good for scavenging free radicals, so it would be important to distinguish among them for establishing quantitative AOC trends. Therefore, it can be anticipated that the predicted trends in AOC may vary depending on the used assay, since they differ in several operational aspects including solvent, pH, and main reaction mechanisms (Table 2).
Considering these aspects altogether, it is not surprising that finding an experimental assay that can be universally used to assess the AOC of any particular compound, accurately and quantitatively so it can be fairly compared with those of other antioxidants, is a very challenging task.
6.2. Theoretical Approaches
Theoretical approaches have their own inherent difficulties as well, mainly associated with the frequent necessity of using simplified models, and to the availability of adequate strategies for including environmental effects, such as those arising from the solvent. Despite of these difficulties, it has been demonstrated that the QM-ORSA protocol can produce values with uncertainties comparable to those arising from experiments (section 5.3.2). Therefore, the data gathered so far from using this protocol is reviewed herein, and to propose possible trends in primary AOC for a large variety of compounds.
To that purpose, the rate coefficients for the reactions of different potential scavengers with 'OOH have been used. This radical has been chosen for several reasons. It is the smallest member of the peroxyl family, and has been proposed to play an essential role in the toxic side effects associated with aerobic respiration, while more information on it is still necesary.[491] ROO' are among those radicals of biological relevance and can be successfully scavenged to retard OS,[492] since their half-lives are long enough to assure that they can be efficiently intercepted bye antioxidants.[493] In addition, from a theoretical point of view, 'OOH is more adequate than other peroxyl radicals -such as CH3OO'- for kinetic calculations because the transition states involving 'OOH has lower multireference character.
It should be noted that, since the molar fraction of HOO' is a constant at a given pH, it is not necessary to include it for establishing trends in AOC provided that the comparisons are performed at the same pH, albeit it would be crucial to reproduce the observable value of the rate coefficient. Therefore the rate coefficients reported in Table 4 do not include the molar fraction of HOO'. However, this can be easily done by multiplying the values in this table by 0.0025 (the molar fraction of HOO' at pH=7.4). Another important point that deserves to be mentioned is that most meaningful quantity for establishing trends in radical scavenging activity would be the product of the rate constants by concentration. However, since information about the concentrations of the different compounds in biological systems are seldom available, the trends proposed here corresponds to the intrinsic ability of the compared compounds for scavenging radicals, i.e., assuming similar concentrations for all the analyzed scavengers.
The calculated data has been divided in two groups, the first one corresponding to reactions in hydrophobic solvents (Table 3), and the second one for the reactions in aqueous solution, at physiological pH (Table 4). This facilitates proposing separated trends for both media, as recommended. The dashed lines in these tables mark the threshold value, thus the compounds above this line are predicted as efficient primary antioxidants while those located below the line are not. It should be noted, however, that the later can still be able of efficiently scavenging free radicals more reactive than 'OOH, such as 'OH, alcoxyl radicals, and halogenated peroxyl radicals. In addition they might be able of protecting against OS as secondary antioxidants, for example by sequestering metal ions.
According to the data gathered so far, the compounds with best primary AOC, in hydrophobic environments, are proposed to be dopamine, canolol, hydroxytyrosol, gallic acid, and piceatannol, in that order (Table 3). On the other hand, the series changes to piceatannol > propyl gallate > Edaravone > fraxetin > ferulic acid > caffeic acid > sesamol > DHLA, in aqueous solution, at pH=7.4 (Table 4). It is also interesting to note that the amount of chemical compounds able of scavenging 'OOH, at least 10 times faster than Trolox, is much larger in aqueous solution than in hydrophobic solvents (25 vs 8 compounds, respectively). Moreover, the rate constants of the best performing antioxidants are higher by about 3 orders of magnitude in aqueous solution than in hydrophobic environments. Accordingly, it may be expected that most of the free radical scavenging processes would take place in the aqueous phase.
Contrary to what it may seem, this is not a bad thing since most of the free radicals present in living organisms, that represent oxidative hazards to biological targets, are polar species that are expected to be present in larger concentrations in hydrophilic regions, compared to hydrophobic ones. In addition, piceatannol is the only of the analyzed compounds that is among the best primary antioxidants in both environments. This makes it a good candidate to be included in dietary supplements designed to reduce oxidative stress. Another strategy could be mixing two, or more compounds, in such a way that at least there is one of the best antioxidants in aqueous solution, and one of the best antioxidants in the hydrophobic phase.
7. Current Challenges and Future Directions
There are several challenges when investigating chemical reactions relevant to oxidative stress using computational strategies. They are mainly related to the accuracy and reliability of the obtained data. First of all, it is necessary to use complex enough models, thus they represent as completely as possible the actual chemical processes. Environmental effects, such as solvent, pH, and potential interfering agents should be taken into account and properly described. At the same time the size of the modeled system cannot be so large that it prevents using reliable levels of theory. All the mechanisms and sites of reactions should be included, as well as all the relevant acid-base (or tautomeric) forms, which makes the investigations in this field quite laborious, especially as the size of the antioxidant molecule increases. In addition, kinetic calculations are frequently needed to get a complete enough picture of these processes. Therefore the electronic structure method of choice should be reliable enough for that purpose. Last, but not least, to establish trends in antioxidant activity all the analyzed compounds should be studied with the same computational protocol to assure that the results are fairly compared.
Oxidative stress and antioxidant protection are complex and manifold phenomena. This review had been mainly focused on chemical processes, related to free radical induced damages and free radical scavenging activities (primary antioxidants) because it is the area which has been most widely explored so far, at molecular levels. However, there are enzymatic reactions that are also relevant in this context. In addition, antioxidant protection may also arise from metal chelation, absorption of ultraviolet radiation, decomposition of hydroperoxide into non-radical species, deactivation of singlet oxygen, or by scavenging oxygen. All these aspects deserve further investigation, and computational techniques may assist on this pursuit. Benchmark studies demonstrating the reliability of computational protocols designed to accurately describe such processes are desirable, as well as investigations on specific systems that allow proposing trends in activity.
Another aspect that remains almost unexplored is the chemical fate of the products, often free radicals, yielded after the primary scavenging activity takes place. In particular it would be crucial to assess their potential oxidative action towards the most relevant biological targets, if any. Investigations properly assessing interactions between antioxidants and frequently used drugs are also of high relevance. Moreover, the ultimate goal in studying antioxidants is to design efficient strategies to inhibit oxidative stress and its deleterious effects, which requires a huge amount of diverse information. Therefore multidisciplinary investigations in this field are highly desirable and increasingly important to achieve such an ambitious goal.
8. Concluding Remarks
OS is a chemical stress that appears in living organisms when the balance between free radicals production and removal is disturbed. It may have very dangerous consequences to human health. Nowadays, an overwhelming amount of evidence has been gathered connecting OS with numerous diseases. Accordingly, it becomes evident that finding efficient strategies to ameliorate OS is crucial to improve the human health status. They can be classified as prevention, protection or repairing strategies.
Prevention can be achieved by avoiding exposure to the free radical sources, or more realistically, using secondary antioxidants able of inhibiting the free radical production, at least for the most damaging of them ('OH). Protection can be exerted by sacrifice molecules, able of reacting with oxidants before they reach biological targets. When prevention and protection are not enough, thus biomolecules are affected by oxidative damage, repairing is the only remaining option. It can be accomplished enzymatically of through the fast removal of transient radicals by natural and synthetic compounds.
Antioxidants are frequently involved in the three kind of strategies for reducing OS, and can be obtained by humans from both exogenous and endogenous sources. Some of the most important requirements for a chemical compound to be considered a good antioxidant are the lack of toxicity, its availability, distribution and concentration, as well as its versatility and ability to rapidly react with free radical, and to cross physiological barriers.
Antioxidants can exert their free radical scavenging activity by a wide variety of reaction mechanisms, including hydrogen atom transfer, single electron transfer, radical adduct formation, proton coupled electron transfer, sequential proton loss electron transfer, sequential proton loss hydrogen transfer, and sequential electron proton transfer.
Oxidative stress and antioxidant protection are very complex processes that can be investigated using both experimental (in vivo or in vitro) and theoretical (in silieo) approaches, albeit combined studies are probably the best choice. The most common computational strategies used to that purpose include those based on intrinsic reactivity, thermochemical, and kinetic data. The latter are, arguably, the one offering the most complete picture of these phenomena from a chemical point of view. They can be used to establish trends in reactivity that help identifying the best antioxidants, and hopefully designing efficient strategies to reduce OS.
There are still numerous challenges associated with theoretical calculations aiming to investigate oxidative stress processes. Albeit there are still several rather unexplored aspects on this topic, significant progress has been made in the last decades in the understanding of the associated chemistry, and further advances are expected in the near future. Computational based strategies might significantly contribute to this wide and complex area of research.
List of Abbreviations
1- MUA: 1-methyluric acid
23- DHBA: 2,3-dihydroxybenzoic acid (2-pyrocatechuic acid or hypogallic acid)
24- DHBA: 2,4-dihydroxybenzoic acid (β-resorcylic acid)
25- DHBA: 2,5-dihydroxybenzoic acid (gentisic acid)
26- DHBA: 2,6-dihydroxybenzoic acid (γ-resorcylic acid) 2c3e-S∫S: 2-center-3-electron bonded sulfur species
2- PSA: 2-propenesulfenic acid
34- DHBA: 3,4-dihydroxybenzoic acid (protocatechuic acid)
35- DHBA: 3,5-dihydroxybenzoic acid (α-resorcylic acid)
3OHM: cyclic 3-hydroxymelatonin
6OHM: 6-hydroxymelatonin
6-OHD: 6-hydroxydaidzein
8-OHD: 8-hydroxydaidzein
Aβ: amyloid β-peptide
ABTS: 2,2'-azinobis(3-ethylbenzthiazoline-6-sulfonic acid
AFMK: N1-acetyl-N2-formyl-5-methoxykynuramine
AMK: N1-acetyl-5-methoxykynuramine
AOC: antioxidant capacity
BDE: bond dissociation energy
BDE: bond dissociation energy
DFT: density functional theory
DFT: density functional theory
DFT: density functional theory
DHCA: dihydrocaffeic acid
DHLA: dyhydrolipoic acid
DNA: deoxyribonucleic acid
DPPH: 2,2-diphenyl-1-picrylhydrazyl
EA: electron affinity
ETE: electron transfer enthalpy
FCR: Folin-Ciocalteu reagent)
FEDAM: full electron donator acceptor map
FR: free radicals
FRAP: ferric reducing antioxidant power
G: guanine
HAT: hydrogen atom transfer mechanism
HF: Hartree-Fock
HOMO: highest occupied molecular orbital
HTE: Hydrogen transfer enthalpies
HWR: Haber-Weiss recombination
IE: first ionization energy
IRC: intrinsic reaction coordinate
IVTST-M: variational transition state theory by mapping
LUMO: lowest unoccupied molecular orbital
NACA: N-acetylcysteine amide
NAS: N-acetylserotonin
NDGA: nordihydroguaiaretic acid
ORAC: oxygen radical absorbance capacity
OS: oxidative stress PA: proton affinity
PCET: proton coupled electron transfer mechanism
PDE: proton dissociation enthalpy
PES: potential energy surface
PTE: proton transfer enthalpy
QM-ORSA: quantum mechanics-based test for overall free radical scavenging activity
RAF: radical adduct formation mechanism
RS': thiyl radicals
RSOH: sulfenic acids
RS(O)2SR: disulfide-S-oxides
s: reaction coordinate
SdPLET: sequential double proton loss electron transfer
SEPT: sequential electron proton transfer mechanism
SET: single electron transfer mechanism
SMD: solvation model based on density
SOMO: single occupied molecular orbital
SPLET: sequential proton loss electron transfer mechanism
SPLHAT: Sequential proton loss hydrogen atom transfer
TEAC: trolox equivalent antioxidant capacity
TRAP: total radical-trapping antioxidant parameter
TST: conventional transition state theory
ZPE: zero point energy
λ: reorganization energy
Acknowledgements
Helpful discussion with Prof. Alvarez-Idaboy is acknowledged. This work was partially supported by project SEP-CONACyT 167-491.
References
1. Sayre, L. M.; Perry, G.; Smith, M. A. Chem. Res. Toxieol. 2008, 21, 172-188. [ Links ]
2. Pacher, P.; Beckman, J. S.; Liaudet, L. Physiol. Rev. 2007, 87, 315-424. [ Links ]
3. Genestra, M. Cell. Signal. 2007, 19, 1807-1819. [ Links ]
4. Valko, M., et al. Int. J. Bioehem. Cell Biol. 2007, 39, 44-84. [ Links ]
5. Valko, M., et al. Chem. Biol. Interaet. 2006, 160, 1-40. [ Links ]
6. Pham-Huy, L. A.; He, H.; Pham-Huy, C. Int. J. Biomed. Sei. 2008, 4, 89-96. [ Links ]
7. Droge, W. Physiol. Rev. 2002, 82, 47-95. [ Links ]
8. Young, I. S.; Woodside, J. V. J. Clin. Pathol. 2001, 54, 176-186. [ Links ]
9. Halliwell, B. Bioehem. Soe. Trans. 2007, 35, 1147-1150. [ Links ]
10. Kaminskyy, V. O.; Zhivotovsky, B. Antioxid. Redox Signal. 2014, 21, 86-102. [ Links ]
11. Yin, H.; Zhu, M. Free Radie. Res. 2012, 46, 959-974. [ Links ]
12. Griffiths, H. R., et al. Bioehem. Soe. Trans. 2011, 39, 1273-1278. [ Links ]
13. Schetter, A. J.; Heegaard, N. H. H.; Harris, C. C. Careinogenesis. 2009, 31, 37-49. [ Links ]
14. Giannapas, M.; Karnis, L.; Dailianis, S. Comp. Bioehem. Physiol. C Toxieol. Pharmaeol. 2012, 155, 182-189. [ Links ]
15. Lantow, M., et al. Radiat. Res. 2006, 165, 88-94. [ Links ]
16. Ren, Y., et al. Int. J. Neurosei. 2015, 125, 555-565. [ Links ]
17. Niatsetskaya, Z. V., et al. J. Neurosei. 2012, 32, 3235-3244. [ Links ]
18. Maeda, H. Caneer Sei. 2013, 104, 779-789. [ Links ]
19. Guha, M., et al. J. Pineal Res. 2007, 43, 372-381. [ Links ]
20. De Luca, C., et al. Toxieol. Ind. Health. 2009, 25, 259-267. [ Links ]
21. Morales-Alamo, D.; Calbet, J. A. L. Free Radie. Res. 2014, 48, 30-42. [ Links ]
22. Benameur, L., et al. Bio-Med. Mater. Eng. 2015, 25, S41-S46. [ Links ]
23. Shiraiwa, M.; Selzle, K.; Põschl, U. Free Radie. Res. 2012, 46, 927-939. [ Links ]
24. Valencia-Islas, N.; Zambrano, A.; Rojas, J. L. J. Chem. Eeol. 2007, 33, 1619-1634. [ Links ]
25. Kamiñski, P.; Kurhalyuk, N.; Szady-Grad, M. Polish J. Environ. Studies. 2007, 16, 555-562. [ Links ]
26. Vejerano, E.; Lomnicki, S.; Dellinger, B. J. Environ. Monit. 2012, 14, 2803-2806. [ Links ]
27. Robinson, E. A.; Johnson, J. D. Mini Rev. Org. Chem. 2011, 8, 401-411. [ Links ]
28. Wang, Y., et al. Tobaeeo Sei. Teehnol. 2014, 47, 47-53. [ Links ]
29. Michail, K., et al. Chem. Res. Toxieol. 2013, 26, 1872-1883. [ Links ]
30. Aleryani, S. L.; Aleryani, R. A.; Al-Akwa, A. A. Drug Test Anal. 2011, 3, 548-551. [ Links ]
31. Narwaley, M., et al. Chem. Res. Toxieol. 2011, 24, 1031-1039. [ Links ]
32. Karadayian, A. G., et al. Neuroseienee. 2015, 304, 47-59. [ Links ]
33. Albano, E. Proe. Nutr. Soe. 2006, 65, 278-290. [ Links ]
34. Spitz, D. R.; Hauer-Jensen, M. Antioxid. Redox Signal. 2014, 20, 1407-1409. [ Links ]
35. Rancan, F., et al. Skin Res. Teehnol. 2014, 20, 182-193. [ Links ]
36. Burlaka, A., et al. Exp. Oneol. 2013, 35, 219-225. [ Links ]
37. Marnett, L. J. Careinogenesis. 1987, 8, 1365-1373. [ Links ]
38. Pryor, W. A. Annu. Rev. Physiol. 1986, VOL. 48, 657-667. [ Links ]
39. León-Carmona, J. R.; Galano, A. J. Phys. Chem. B. 2011, 115, 4538-4546. [ Links ]
40. Galano, A. J. Phys. Chem. B. 2007, 111, 12898-12908. [ Links ]
41. Martínez, A.; Vargas, R.; Galano, A. J. Phys. Chem. B. 2009, 113, 12113-12120. [ Links ]
42. Martínez, A.; Vargas, R.; Galano, A. Theor. Chem. Aee. 2010, 127, 595-603. [ Links ]
43. Martínez, A.; Galano, A. Journal of Physieal Chemistry C. 2010, 114, 8184-8191. [ Links ]
44. Brunelli, L.; Crow, J. P.; Beckman, J. S. Areh. Bioehem. Biophys. 1995, 316, 327-334. [ Links ]
45. Squadrito, G. L.; Pryor, W. A. Free Radieal Biology and Medieine. 1998, 25, 392-403. [ Links ]
46. Radi, R., et al. Free Radieal Biology and Medieine. 2001, 30, 463-488. [ Links ]
47. Douki, T.; Cadet, J. Free RadiealReseareh. 1996, 24, 369-380. [ Links ]
48. Wiseman, H.; Halliwell, B. Bioehem. J. 1996, 313, 17-29. [ Links ]
49. Koppal, T., et al. J. Neuroehem. 1999, 72, 310-317. [ Links ]
50. Forni, L. G., et al. Chemieo-Biologieal Interaetions. 1983, 45, 171-177. [ Links ]
51. Prütz, W. A., et al. Areh. Bioehem. Biophys. 1985, 243, 125-134. [ Links ]
52. Abedinzadeh, Z. Can. J. Physiol. Pharmaeol. 2001 , 79, 166-170. [ Links ]
53. Giles, G. I.; Tasker, K. M.; Jacob, C. Free Radie. Biol. Med. 2001, 31, 1279-1283. [ Links ]
54. Giles, G. I.; Tasker, K. M.; Jacob, C. Gen. Physiol. Biophys. 2002, 21, 65-72. [ Links ]
55. Mishanina, T. V.; Libiad, M.; Banerjee, R. Nat. Chem. Biol. 2015, 11, 457-464. [ Links ]
56. Wenska, G., et al. J. Phys. Chem. B. 2008, 112, 10045-10053. [ Links ]
57. Glass, R. S., et al. Tetrahedron Lett. 1992, 33, 1721-1724. [ Links ]
58. Asmus, K. D. Methods Enzymol. 1990, 186, 168-180. [ Links ]
59. Bonifacio, M.; Asmus, K. D. J. Org. Chem. 1986, 51, 1216-1222. [ Links ]
60. Gõbl, M.; Bonifacio, M.; Asmus, K. D. J. Am. Chem. Soe. 1984, 106, 5984-5988. [ Links ]
61. Fourré, I.; Silvi, B. Heteroat. Chem. 2007, 18, 135-160. [ Links ]
62. McKee, M. L. J. Phys. Chem. 1992, 96, 1675-1679. [ Links ]
63. Brunelle, P.; Rauk, A. J. Phys. Chem. A. 2004, 108, 11032-11041. [ Links ]
64. Schõneich, C., et al. J. Am. Chem. Soe. 2003, 125, 13700-13713. [ Links ]
65. Wiberg, K. B.; Petersson, G. A. J. Phys. Chem. A. 2014, 118, 2353-2359. [ Links ]
66. Gerschman, R., et al. Seienee. 1954, 119, 623-626.
67. MacNee, W. Eur. J. Pharmaeol. 2001, 429, 195-207. [ Links ]
68. Caramori, G.; Papi, A. Thorax. 2004, 59, 170-173. [ Links ]
69. Guo, R. F.; Ward, P. A. Antioxid. Redox Signal. 2007, 9, 19912002. [ Links ]
70. Hoshino, Y.; Mishima, M. Antioxid. Redox Signal. 2008, 10, 701-704. [ Links ]
71. Shadab, M., et al. J. Clin. DiagnostieRes. 2014, 8, BC11-BC13. [ Links ]
72. Zhao, W., et al. J. Surg. Res. 2014, 187, 542-552. [ Links ]
73. Ghasemzadeh, N., et al. Hypertension. 2014, 63, 1270-1275. [ Links ]
74. Cheresh, P., et al. Bioehim. Biophys. Aeta. 2013, 1832, 10281040. [ Links ]
75. Pandey, R., et al. J. Clin. Diagn. Res. 2013, 7, 580-588. [ Links ]
76. Lanzetti, M., et al. Free Radie. Biol. Med. 2012, 53, 1993-2001. [ Links ]
77. Zuo, L., et al. Frontiers in Biology. 2012, 7, 506-513. [ Links ]
78. Kratzer, E., et al. Am. J. Respir. Cell Mol. Biol. 2012, 47, 688697. [ Links ]
79. Galle, J. Nephrol. Dial. Transplant. 2001, 16, 2135-2137. [ Links ]
80. Yang, S. K., et al. Ren. Fail. 2014, 36, 313-320. [ Links ]
81. Aruna, G.; Ambika Devi, K. Int. J. Pharma Bio Sei. 2014, 5, B127-B133. [ Links ]
82. Araujo, A. M., et al. Am. J. Trop. Med. Hyg. 2014, 90, 719-723. [ Links ]
83. Sung, C. C., et al. Oxid. Med. Cell. Longev. 2013, 2013, 301982. [ Links ]
84. Small, D. M., et al. Nephron Exp. Nephrol. 2013, 122, 123-130. [ Links ]
85. Ozbek, E. Int. J. Nephrol. 2012, 2012, 465897. [ Links ]
86. Massy, Z. A.; Stenvinkel, P.; Drueke, T. B. Semin. Dial. 2009, 22, 405-408. [ Links ]
87. Beatty, S., et al. Surv. Ophthalmol. 2000, 45, 115-134. [ Links ]
88. Rosenstein, R. E., et al. J. Pineal Res. 2010, 49, 1-13. [ Links ]
89. Uchino, Y., et al. Cornea. 2012, 31, S63-S67. [ Links ]
90. Kaur, J., et al. J. Clin. Diagn. Res. 2012, 6, 1629-1632. [ Links ]
91. Wakamatsu, T. H., et al. Mol. Vis. 2010, 16, 2465-2475. [ Links ]
92. Dogru, M., et al. Cornea. 2009, 28, S70-S74. [ Links ]
93. Dong, A., et al. J. Cell. Physiol. 2009, 219, 544-552. [ Links ]
94. Mahajan, A.; Tandon, V. R. J. Indian Rheumatol. Assoe. 2004, 12, 139-142. [ Links ]
95. Veselinovic, M., et al. Mol. Cell. Bioehem. 2014, 391, 225-232. [ Links ]
96. Kundu, S., et al. Free Radieal Res. 2012, 46, 1482-1489. [ Links ]
97. Wruck, C. J., et al. Ann. Rheum. Dis. 2011, 70, 844-850. [ Links ]
98. Vasanthi, P.; Nalini, G.; Rajasekhar, G. Int. J. Rheum. Dis. 2009, 12, 29-33. [ Links ]
99. Myatt, L. J. Physiol. 2006, 572, 25-30. [ Links ]
100. Braekke, K.; Harsem, N. K.; Staff, A. C. Pediatr. Res. 2006, 60, 560-564. [ Links ]
101. Biri, A., et al. Gyneeol. Obstet. Invest. 2007, 64, 187-192. [ Links ]
102. Hracsko, Z., et al. Redox Rep. 2008, 13, 11-16. [ Links ]
103. Gitto, E., et al. J. Pineal Res. 2009, 46, 128-139. [ Links ]
104. Mert, I., et al. J. Obstet. Gynaeeol. Res. 2012, 38, 658-664. [ Links ]
105. Valko, M., et al. Mol. Cell. Bioehem. 2004, 266, 37-56. [ Links ]
106. Boyd, N. F.; McGuire, V. Free Radie. Biol. Med. 1991, 10, 185-190. [ Links ]
107. Nelson, R. L. Free Radie. Biol. Med. 1992, 12, 161-168. [ Links ]
108. Knekt, P., et al. Int. J. Caneer. 1994, 56, 379-382. [ Links ]
109. Omenn, G. S., et al. N. Engl. J. Med. 1996, 334, 1150-1155. [ Links ]
110. Willcox, J. K.; Ash, S. L.; Catignani, G. L. Crit. Rev. Food Sei. Nutr. 2004, 44, 275-295. [ Links ]
111. Wang, J., et al. Oneol. Lett. 2014, 7, 1159-1164. [ Links ]
112. Granados-Principal, S., et al. Bioehem. Pharmaeol. 2014, 90, 2533. [ Links ]
113. Tekiner-Gulbas, B.; Westwell, A. D.; Suzen, S. Curr. Med. Chem. 2013, 20, 4451-4459. [ Links ]
114. Sayre, L. M.; Smith, M. A.; Perry, G. Curr. Med. Chem. 2001, 8, 721-738. [ Links ]
115. Butterfield, D. A., et al. Bioehem. Biophys. Res. Commun. 1994, 200, 710-715. [ Links ]
116. Hensley, K., et al. Proe. Natl. Aead. Sei. U. S. A. 1994, 91, 3270-3274. [ Links ]
117. Butterfield, D. A., et al. Life Sei. 1996, 58, 217-228. [ Links ]
118. Butterfield, D. A. Chem. Res. Toxieol. 1997, 10, 495-506. [ Links ]
119. Mattson, M. P. Nature. 2004, 430, 631-639. [ Links ]
120. Christen, Y. Am. J. Clin. Nutr. 2000, 71, 621S-629S. [ Links ]
121. Halliwell, B. Drugs Aging. 2001, 18, 685-716. [ Links ]
122. Butterfield, D. A. Free Radie. Res. 2002, 36, 1307-1313. [ Links ]
123. Cruz-Sanchez, F. F., et al. J. Neurol. Sei. 2010, 299, 163-167. [ Links ]
124. Bonda, D. J., et al. Neuropharmaeology. 2010, 59, 290-294. [ Links ]
125. Cai, Z.; Zhao, B.; Ratka, A. NeuromoleeularMed. 2011, 13, 223250. [ Links ]
126. Chen, Z.; Zhong, C. Neurosei. Bull. 2014, 30, 271-281. [ Links ]
127. Eskici, G.; Axelsen, P. H. Bioehemistry. 2012, 51, 6289-6311. [ Links ]
128. López, N., et al. J. Alzheimers Dis. 2013, 33, 823-829. [ Links ]
129. Meraz-Ríos, M. A., et al. Oxid. Med. Cell. Longev. 2014, 2014, 375-968. [ Links ]
130. Pimentel, C., et al. Oxid.Med. Cell. Longev. 2012, 2012, 132146. [ Links ]
131. Pocernich, C. B., et al. Curr. Alzheimer Res. 2011, 8, 452-469. [ Links ]
132. Pohanka, M. Curr. Med. Chem. 2014, 21, 356-364. [ Links ]
133. Rosini, M., et al. J. Med. Chem. 2014, 57, 2821-2831. [ Links ]
134. Schrag, M., et al. Neurobiol. Dis. 2013, 59, 100-110. [ Links ]
135. Torres, L. L., et al. J. Alzheimers Dis. 2011, 26, 59-68. [ Links ]
136. Yana, M. H.; Wang, X.; Zhu, X. Free Radieal Bio. Med. 2013, 62, 90-101. [ Links ]
137. Zhao, Y.; Zhao, B. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [ Links ]
138. Janero, D. R. Free Radie. Biol. Med. 1991, 11, 129-144. [ Links ]
139. Steinberg, D. Circulation. 1991, 84, 1420-1425. [ Links ]
140. Riemersma, R. A., et al. Laneet. 1991, 337, 1-5.
141. Salonen, J. T., et al. Cireulation. 1992, 86, 803-811. [ Links ]
142. Street, D. A., et al. Cireulation. 1994, 90, 1154-1161.
143. Hodis, H. N., et al. J. Am. Med. Assoe. 1995, 273, 1849-1854. [ Links ]
144. Kushi, L. H., et al. N. Engl. J. Med. 1996, 334, 1156-1162. [ Links ]
145. Stephens, N. G., et al. Laneet. 1996, 347, 781-786.
146. Panasenko, O. M., et al. Free Radieal Bio. Med. 1991, 10, 137148. [ Links ]
147. Matsuda, M.; Shimomura, I. Rev. Endoer. Metab. Disord. 2014, 15, 1-10. [ Links ]
148. Eren, E., et al. Redox report : eommunieations in free radieal researeh. 2014, 19, 34-39.
149. Al-Aubaidy, H. A.; Jelinek, H. F. Redox Rep. 2014, 19, 87-91. [ Links ]
150. Serban, C.; Dragan, S. Curr. Pharm. Design. 2014, 20, 585-600. [ Links ]
151. Zampetaki, A.; Dudek, K.; Mayr, M. Free Radie. Biol. Med. 2013, 64, 69-77. [ Links ]
152. Cadet, J.; Douki, T.; Ravanat, J.-L. Aee. Chem. Res. 2008, 41, 1075-1083. [ Links ]
153. Kawanishi, S.; Hiraku, Y.; Oikawa, S. Mutat. Res. 2001, 488, 6576. [ Links ]
154. Sevilla, M. D.; Besler, B.; Colson, A.-O. J. Phys. Chem. 1995, 99, 1060-1063. [ Links ]
155. Seidel, C. A. M.; Schulz, A.; Sauer, M. H. M. J. Phys. Chem. 1996, 100, 5541-5553. [ Links ]
156. Sugiyama, H.; Saito, I. J. Am. Chem. Soe. 1996, 118, 7063-7068. [ Links ]
157. Melvin, T., et al. J. Am. Chem. Soe. 1996, 118, 10031-10036. [ Links ]
158. Wetmore, S. D.; Boyd, R. J.; Eriksson, L. A. Chem. Phys. Lett. 2000, 322, 129-135. [ Links ]
159. Langmaier, J., et al. J. Phys. Chem. B. 2004, 108, 15896-15899. [ Links ]
160. Burrows, C. J.; Muller, J. G. Chem. Rev. 1998, 98, 1109-1152. [ Links ]
161. Angelov, D., et al. J. Am. Chem. Soe. 1997, 119, 11373-11380. [ Links ]
162. Melvin, T., et al. Photoehem. Photobiol. 1997, 65, 660-665. [ Links ]
163. Melvin, T., et al. J. Chem. Soe., Chem. Commun. 1995, 653-654. [ Links ]
164. Saito, I., et al. J. Am. Chem. Soe. 1995, 117, 6406-6407. [ Links ]
165. Breslin, D. T.; Schuster, G. B. J. Am. Chem. Soe. 1996, 118, 2311-2319. [ Links ]
166. Stemp, E. D. A.; Arkin, M. R.; Barton, J. K. J. Am. Chem. Soe. 1997, 119, 2921-2925. [ Links ]
167. Armitage, B., et al. Proe. Natl. Aead. Sei. U. S. A. 1997, 94, 12320-12325. [ Links ]
168. Saito, I., et al. J. Am. Chem. Soe. 1998, 120, 12686-12687. [ Links ]
169. Yoshioka, Y., et al. J. Am. Chem. Soe. 1999, 121, 8712-8719. [ Links ]
170. Steenken, S. Chem. Rev. 1989, 89, 503-520. [ Links ]
171. Galano, A.; Alvarez-Idaboy, J. R. PCCP. 2012, 14, 12476-12484. [ Links ]
172. Pratviel, G.; Bernadou, J.; Meunier, B. Angew. Chem. Int. Ed. 1995, 34, 746-769. [ Links ]
173. Pogozelski, W. K.; Tullius, T. D. Chem. Rev. 1998, 98, 10891108. [ Links ]
174. Shukla, L. I., et al. Radiat. Res. 2004, 161, 582-590. [ Links ]
175. Dedon, P. C. Chem. Res. Toxieol. 2007, 21, 206-219. [ Links ]
176. Kim, J.; Kreller, C. R.; Greenberg, M. M. J. Org. Chem. 2005, 70, 8122-8129. [ Links ]
177. Tronche, C.; Goodman, B. K.; Greenberg, M. M. Chem. Biol. 1998, 5, 263-271. [ Links ]
178. Borrego, S., et al. Int. J. Mol. Sei. 2013, 14, 3467-3486. [ Links ]
179. De Iuliis, G. N., et al. Biol. Reprod. 2009, 81, 517-524. [ Links ]
180. Mangal, D., et al. Chem. Res. Toxieol. 2009, 22, 788-797. [ Links ]
181. Tope, A. M.; Panemangalore, M. J. Environ. Sei. Health, Pt. B: Pestie., Food Contam., Agrie. Wastes. 2007, 42, 151-155.
182. Chen, S. S., et al. J. Urol. 2004, 172, 1418-1421. [ Links ]
183. Tarng, D. C., et al. Am. J. Kidney Dis. 2000, 36, 934-944. [ Links ]
184. Kasai, H. Mutat. Res. - Rev. Mut. Res. 1997, 387, 147-163. [ Links ]
185. Mozziconacci, O.; Kerwin, B. A.; Schõneich, C. Chem. Res. Tox-ieol. 2010, 23, 1310-1312. [ Links ]
186. Lund, M. N., et al. Bioehem. J. 2008, 410, 565-574. [ Links ]
187. Lardinois, O. M.; Ortiz De Montellano, P. R. J. Biol. Chem. 2001, 276, 23186-23191. [ Links ]
188. Lardinois, O. M.; Medzihradszky, K. F.; Ortiz De Montellano, P. R. J. Biol. Chem. 1999, 274, 35441-35448. [ Links ]
189. Hands, S., et al. J. Biol. Chem. 2011, 286, 44512-44520. [ Links ]
190. Hawkins, C. L.; Pattison, D. I.; Davies, M. J. Bioehem. J. 2002, 365, 605-615. [ Links ]
191. Hawkins, C. L.; Davies, M. J. Bioehem. J. 1999, 340, 539-548. [ Links ]
192. Headlam, H. A., et al. Chem. Res. Toxieol. 2000, 13, 1087-1095. [ Links ]
193. Grune, T., et al. J. Biol. Chem. 1995, 270, 2344-2351. [ Links ]
194. Pacifici, R. E.; Kono, Y.; Davies, K. J. A. J. Biol. Chem. 1993, 268, 15405-15411. [ Links ]
195. Silakov, A., et al. J. Am. Chem. Soe. 2014, 136, 8221-8228. [ Links ]
196. Decroos, C., et al. Chem. Res. Toxieol. 2014, 27, 627-636. [ Links ]
197. Chung, T. W., et al. J. Am. Soe. Mass Speetrom. 2012, 23, 13361350. [ Links ]
198. Pattison, D. I.; Davies, M. J. Bioehemistry. 2004, 43, 4799-4809. [ Links ]
199. Stadtman, E. R.; Levine, R. L. AminoAeids. 2003, 25, 207-218. [ Links ]
200. Filipe, P., et al. Bioehemistry. 2002, 41, 11057-11064. [ Links ]
201. Elias, R. J.; McClements, D. J.; Decker, E. A. J. Agrie. Food Chem. 2005, 53, 10248-10253. [ Links ]
202. Alvarez, B.; Radi, R. Amino Aeids. 2003, 25, 295-311. [ Links ]
203. Nadal, R. C.; Rigby, S. E. J.; Viles, J. H. Bioehemistry. 2008, 47, 11653-11664. [ Links ]
204. Nadal, R. C., et al. Free Radieal Bio. Med. 2007, 42, 79-89. [ Links ]
205. Kim, N. H., et al. Mol. Cells. 2006, 22, 220-227. [ Links ]
206. Arenas, A., et al. Chem. Res. Toxieol. 2013, 26, 67-77. [ Links ]
207. Beal, J. L.; Foster, S. B.; Ashby, M. T. Bioehemistry. 2009, 48, 11142-11148. [ Links ]
208. Estevam, M. L., et al. J. Biol. Chem. 2004, 279, 39214-39222. [ Links ]
209. Hawkins, C. L.; Davies, M. J. Free Radie. Biol. Med. 2005, 39, 900-912. [ Links ]
210. Mozziconacci, O., et al. Free Radieal Bio. Med. 2007, 43, 229240. [ Links ]
211. Galletti, P., et al. FEBS J. 2007, 274, 5263-5277. [ Links ]
212. Chatgilialoglu, C., et al. J. Proteomies. 2011, 74, 2264-2273. [ Links ]
213. Moosmann, B. Exp. Gerontol. 2011, 46, 164-169. [ Links ]
214. Kwon, D. Y., et al. J. Nutr. 2009, 139, 63-68. [ Links ]
215. Tsakiris, S., et al. Pharmaeol. Res. 2006, 53, 386-390. [ Links ]
216. Miller, B. L.; Williams, T. D.; Schõneich, C. J. Am. Chem. Soe. 1996, 118, 11014-11025. [ Links ]
217. Naslund, J., et al. Proe. Natl. Aead. Sei. U. S. A. 1994, 91, 83788382. [ Links ]
218. Bhatia, S., et al. Bioehem. J. 2012, 442, 713-721. [ Links ]
219. Nagy, P.; Kettle, A. J.; Winterbourn, C. C. Free Radie. Biol. Med. 2010, 49, 792-799. [ Links ]
220. Kontush, A.; Chapman, M. J. Curr. Opin. Lipidol. 2010, 21, 312318. [ Links ]
221. Thariat, J., et al. Bioehimie. 2008, 90, 1442-1451. [ Links ]
222. Nakao, L. S., et al. FEBS Lett. 2003, 547, 87-91. [ Links ]
223. Fay, D. S., et al. J. Neuroehem. 1998, 71, 1616-1625. [ Links ]
224. Clementi, M. E., et al. Bioehem. Biophys. Res. Commun. 2006, 342, 206-213. [ Links ]
225. Ali, F. E., et al. J. Pept. Sei. 2005, 11, 353-360. [ Links ]
226. Schõneich, C. Bioehim. Biophys. Aeta. 2005, 1703, 111-119. [ Links ]
227. Clementi, M. E., et al. Int. J. Bioehem. Cell Biol. 2004, 36, 20662076. [ Links ]
228. Misiti, F., et al. Neuroseienee. 2004, 126, 297-303. [ Links ]
229. Butterfield, D. A.; Kanski, J. Peptides. 2002, 23, 1299-1309. [ Links ]
230. Varadarajan, S., et al. J. Struet. Biol. 2000, 130, 184-208. [ Links ]
231. Yatin, S. M., et al. Neurobiol. Aging. 1999, 20, 325-330. [ Links ]
232. Pike, C. J., et al. J. Neuroehem. 1995, 64, 253-265. [ Links ]
233. Varadarajan, S., et al. Brain Res. Bull. 1999, 50, 133-141. [ Links ]
234. Francisco-Marquez, M.; Galano, A. J. Phys. Chem. B. 2009, 113, 4947-4952. [ Links ]
235. Boyd-Kimball, D., et al. Peptides. 2005, 26, 665-673. [ Links ]
236. Hawkins, C. L.; Davies, M. J. J. Chem. Soe. Perk. Trans. 2. 1998, 2617-2622.
237. Chan, B., et al. J. Org. Chem. 2012, 77, 9807-9812. [ Links ]
238. Trouillas, P.; Bergès, J.; Houée-Lévin, C. Int. J. Quantum Chem. 2011, 111, 1143-1151. [ Links ]
239. Pryor, W. A. Free Radieal Bio. Med. 1988, 4, 219-223. [ Links ]
240. Vijayalaxmi, et al. Int. J. Radiat. Oneol. Biol. Phys. 2004, 59, 639-653. [ Links ]
241. Candeias, L. P.; Steenken, S. Chem. - Eur. J. 2000, 6, 475-484. [ Links ]
242. Chatgilialoglu, C., et al. Angew. Chem. Int. Ed. Engl. 2009, 48, 2214-2217. [ Links ]
243. Galano, A.; Alvarez-Idaboy, J. R. Org. Lett. 2009, 11, 5114-5117. [ Links ]
244. Weinstein, J.; Bielski, B. H. J. J. Am. Chem. Soe. 1979, 101, 5862. [ Links ]
245. Remucal, C. K.; Sedlak, D. L., in: ACS Symp. Ser., Vol. 1071, 2011, 177-197. [ Links ]
246. Salgado, P., et al. Journal of the Chilean ChemiealSoeiety. 2013, 58, 2096-2101. [ Links ]
247. Bokare, A. D.; Choi, W. Environ. Sei. Teehnol. 2010, 44, 72327237. [ Links ]
248. Yao, B., et al., in: Adv. Mater. Res., Vol. 1010-1012, 2014, 84-87. [ Links ]
249. Bang, S., et al. Angew. Chem. Int. Ed. Engl. 2014, 53, 7843-7847. [ Links ]
250. Silaghi-Dumitrescu, R. Areh. Bioehem. Biophys. 2004, 424, 137140. [ Links ]
251. Sutton, H. C. Journal ofthe Chemieal Soeiety, Faraday Transae-tions 1: Physieal Chemistry in Condensed Phases. 1989, 85, 883-893. [ Links ]
252. Hong, J.; Schõneich, C. Free Radie. Biol. Med. 2001, 31, 14321441. [ Links ]
253. Berthon, G. AgentsAetions. 1993, 39, 210-217. [ Links ]
254. Galano, A.; Francisco Marquez, M.; Pérez-González, A. Chem. Res. Toxieol. 2014, 27, 904-918. [ Links ]
255. Galano, A., et al. J. Pineal Res. 2014, [ Links ]
256. Zheng, R., et al. Chem. Soe. Rev. 2010, 39, 2827-2834. [ Links ]
257. Loeb, L. A. CaneerRes. 1989, 49, 5489-5496. [ Links ]
258. Shuryak, I.; Brenner, D. J. J. Theor. Biol. 2009, 261, 305-317. [ Links ]
259. Culard, F., et al. J. Mol. Biol. 2003, 328, 1185-1195. [ Links ]
260. Jolivet, E., et al. Mol. Mierobiol. 2006, 59, 338-349. [ Links ]
261. Kowalczyk, A.; Serafin, E.; Puchala, M. Int. J. RadiatBiol. 2008, 84, 15-22. [ Links ]
262. Liu, Y., et al. Proe. Natl. Aead. Sei. U. S. A. 2003, 100, 41914196. [ Links ]
263. Gorbunova, V., et al. Nueleie Aeids Res. 2007, 35, 7466-7474. [ Links ]
264. Schulte-Frohlinde, D.; Behrens, G.; Onal, A. Int. J. Radiat Biol. 1986, 50, 103-110. [ Links ]
265. Jaruga, P.; Dizdaroglu, M. Nueleie Aeids Res. 1996, 24, 13891394. [ Links ]
266. Yakes, F. M.; Van Houten, B. Proe. Natl. Aead. Sei. USA. 1997, 94, 514-519. [ Links ]
267. Anderson, R. F., et al. Free Radie. Res. 2000, 33, 91-103. [ Links ]
268. Anderson, R. F., et al. Careinogenesis. 2001, 22, 1189-1193. [ Links ]
269. Fu, H., et al. J. Radiat. Res. 2008, 49, 609-614. [ Links ]
270. Milligan, J. R., et al. J. Am. Chem. Soe. 2004, 126, 1682-1687. [ Links ]
271. Jiang, Y., et al. Radiat. Phys. Chem. 1999, 54, 349-353. [ Links ]
272. Ly, A., et al. Bioehemistry. 2004, 43, 9098-9104. [ Links ]
273. Jovanovic, S. V.; Simic, M. G. Bioehim. Biophys. Aeta. 1989, 1008, 39-44. [ Links ]
274. Anderson, R. F.; Harris, T. A. Free Radie. Res. 2003, 37, 11311136. [ Links ]
275. Ly, A., et al. Org. Biomol. Chem. 2005, 3, 917-923. [ Links ]
276. Pellmar, T. C.; Roney, D.; Lepinski, D. L. Brain Res. 1992, 583, 194-200. [ Links ]
277. Pujari, G., et al. Mutat. Res. 2009, 675, 23-28. [ Links ]
278. Kirsch, M., et al. Chem. Eur. J. 2001, 7, 3313-3320. [ Links ]
279. Gebicki, J. M., et al. Amino Aeids. 2010, 39, 1131-1137. [ Links ]
280. Alvarez-Idaboy, J. R.; Galano, A. J. Phys. Chem. B. 2012, 116, 9316-9325. [ Links ]
281. Nauser, T.; Koppenol, W. H.; Schõneich, C. Free Radie. Biol. Med. 2014, 80, 158-163. [ Links ]
282. Hill, B. G.; Bhatnagar, A. J. Mol. Cell. Cardiol. 2012, 52, 559567. [ Links ]
283. Klatt, P.; Lamas, S. Eur. J. Bioehem. 2000, 267, 4928-4944. [ Links ]
284. Adachi, T.; Schõneich, C.; Cohen, R. A. Drug Diseov. Today Dis. Meeh. 2005, 2, 39-46. [ Links ]
285. Rao, A. V.; Agarwal, S. Nutr. Res. 1999, 19, 305-323. [ Links ]
286. Halliwell, B., et al. Crit. Rev. Food Sei. Nutr. 1995, 35, 7-20. [ Links ]
287. Halliwell, B.; Whiteman, M. Br. J. Pharmaeol. 2004, 142, 231255. [ Links ]
288. Chaiyasit, W., et al. Crit. Rev. Food Sei. Nutr. 2007, 47, 299-317. [ Links ]
289. Rose, R. C.; Bode, A. M. FASEB J. 1993, 7, 1135-1142. [ Links ]
290. Galano, A.; Francisco-Marquez, M. J. Phys. Chem. B. 2009, 113, 11338-11345. [ Links ]
291. Mortensen, A., et al. FEBS Lett. 1997, 418, 91-97. [ Links ]
292. Liebler, D. C.; McClure, T. D. Chem. Res. Toxieol. 1996, 9, 8-11. [ Links ]
293. Mortensen, A. Free Radie. Res. 2002, 36, 211-216. [ Links ]
294. Joshi, R., et al. Free Radie. Res. 2012, 46, 11-20. [ Links ]
295. Hata, K., et al. J. Radiat. Res. 2011, 52, 15-23. [ Links ]
296. Pérez-González, A.; Galano, A. J. Phys. Chem. B. 2011, 115, 1306-1314. [ Links ]
297. Galano, A. PCCP. 2011, 13, 7178-7188. [ Links ]
298. Galano, A.; Tan, D. X.; Reiter, R. J. J. Pineal Res. 2013, 54, 245257. [ Links ]
299. Galano, A.; Tan, D. X.; Reiter, R. J. RSC Advanees. 2014, 4, 5220-5227. [ Links ]
300. Dhiman, S. B.; Kamat, J. P.; Naik, D. B. Chem. Biol. Interaet. 2009, 182, 119-127. [ Links ]
301. Sakurai, K., et al. Free Radie. Res. 2004, 38, 487-494. [ Links ]
302. Tamba, M.; Torreggiani, A. Int. J. Radiat Biol. 1999, 75, 11771188. [ Links ]
303. Barzegar, A. Food Chem. 2012, 135, 1369-1376. [ Links ]
304. Touriño, S., et al. Chem. Res. Toxieol. 2008, 21, 696-704. [ Links ]
305. Pérez-González, A.; Galano, A. J. Phys. Chem. B. 2012, 116, 1180-1188. [ Links ]
306. Nakanishi, I., et al. Chem. Res. Toxieol. 2004, 17, 26-31. [ Links ]
307. Nakanishi, I., et al. Chem. Lett. 2007, 36, 1276-1277. [ Links ]
308. Hill, T. J., et al. J. Am. Chem. Soe. 1995, 117, 8322-8326. [ Links ]
309. Packer, J. E., et al. Bioehem. Biophys. Res. Commun. 1981, 98, 901-906. [ Links ]
310. Everett, S. A., et al. Bioehem. Soe. Trans. 1995, 23, 230S. [ Links ]
311. Galano, A.; Vargas, R.; Martínez, A. PCCP. 2010, 12, 193-200. [ Links ]
312. Martínez, A.; Hernández-Marin, E.; Galano, A. Food and Funetion. 2012, 3, 442-450. [ Links ]
313. Sueishi, Y., et al. Food Chem. 2011, 129, 866-870. [ Links ]
314. Cao, L., et al. Anal. Methods. 2014, 6, 7149-7153. [ Links ]
315. Mendoza-Wilson, A. M.; Castro-Arredondo, S. I.; Balandrán-Quintana, R. R. Food Chem. 2014, 161, 155-161. [ Links ]
316. Li, X., et al. Chem. Biol. Interaet. 2014, 219, 221-228. [ Links ]
317. Wang, G., et al. Food Chem. 2014, 171, 89-97. [ Links ]
318. Praveena, R., et al. J. Mol. Struet. 2014, 1061, 114-123. [ Links ]
319. Mikulski, D.; Eder, K.; Molski, M. J. Theor. Comput. Chem. 2014, 13, [ Links ]
320. Galano, A.; Martínez, A. J. Phys. Chem. B. 2012, 116, 12001208. [ Links ]
321. Martínez, A.; Galano, A.; Vargas, R. J. Phys. Chem. B. 2011, 115, 12591-12598. [ Links ]
322. Dimitric Markovic, J. M., et al. RSC Advanees. 2014, 4, 32228-32236. [ Links ]
323. Mazzone, G.; Toscano, M.; Russo, N. J. Agrie. Food Chem. 2013, 61, 9650-9657. [ Links ]
324. Xue, Y., et al. Comput. Theor. Chem. 2012, 982, 74-83. [ Links ]
325. Castañeda-Arriaga, R.; Alvarez-Idaboy, J. R. J. Chem. Inf. Model. 2014, 54, 1642-1652. [ Links ]
326. Farmanzadeh, D.; Najafi, M. Bull. Chem. Soe. Jpn. 2013, 86, 1041-1050. [ Links ]
327. Galano, A.; Alvarez-Idaboy, J. R. RSC Adv. 2011, 1, 1763-1771. [ Links ]
328. Galano, A. Theor. Chem. Aee. 2011, 130, 51-60. [ Links ]
329. Jeremie, S., et al. Comput. Theor. Chem. 2014, 1047, 15-21. [ Links ]
330. Xue, Y., et al. Food Chem. 2014, 151, 198-206. [ Links ]
331. Medina, M. E.; Galano, A.; Alvarez-Idaboy, J. R. Phys. Chem. Chem. Phys. 2014, 16, 1197-1207. [ Links ]
332. Markovic, Z., et al. Monatsh. Chem. 2014, 145, 953-962. [ Links ]
333. Pérez-González, A.; Galano, A.; Alvarez-Idaboy, J. R. New J. Chem. 2014, 38, 2639-2652. [ Links ]
334. Medina, M. E.; Iuga, C.; Álvarez-Idaboy, J. R. RSC Adv. 2014, 4, 52920-52932. [ Links ]
335. Caicedo, C., et al. RSC Adv. 2014, 4, 38918-38930. [ Links ]
336. Iuga, C.; Alvarez-Idaboy, J. R.; Russo, N. J. Org. Chem. 2012, 77, 3868-3877. [ Links ]
337. Cordova-Gomez, M.; Galano, A.; Alvarez-Idaboy, J. R. RSC Adv. 2013, 3, 20209-20218. [ Links ]
338. Shang, Y. J., et al. J. Org. Chem. 2009, 74, 5025-5031. [ Links ]
339. Xue, Y., et al. J. Phys. Org. Chem. 2013, 26, 240-248. [ Links ]
340. Markovic, Z., et al. Food Chem. 2012, 135, 2070-2077. [ Links ]
341. Di Meo, F., et al. J. Phys. Chem. A. 2013, 117, 2082-2092. [ Links ]
342. Erdemgil, F. Z., et al. Talanta. 2007, 72, 489-496. [ Links ]
343. Galano, A.; Alvarez-Idaboy, J. R. J. Comput. Chem. 2013, 34, 2430-2445. [ Links ]
344. Mayer, J. M., et al. J. Am. Chem. Soe. 2002, 124, 11142-11147. [ Links ]
345. DiLabio, G. A.; Ingold, K. U. J. Am. Chem. Soe. 2005, 127, 6693-6699. [ Links ]
346. Sirjoosingh, A.; Hammes-Schiffer, S. J. Phys. Chem. A. 2011, 115, 2367-2377. [ Links ]
347. Tishchenko, O., et al. J. Am. Chem. Soe. 2008, 130, 7000-7010. [ Links ]
348. DiLabio, G. A.; Johnson, E. R. J. Am. Chem. Soe. 2007, 129, 6199-6203. [ Links ]
349. Amic, A., et al. Food Chem. 2014, 152, 578-585. [ Links ]
350. Barzegar, A. PLoS One. 2012, 7. [ Links ]
351. Nakayama, T.; Uno, B. Chem. Lett. 2010, 39, 162-164. [ Links ]
352. Inagaki, T.; Yamamoto, T. J. Phys. Chem. B. 2014, 118, 937-950. [ Links ]
353. Li, M., et al. Int. J. Quantum Chem. 2013, 113, 966-974. [ Links ]
354. Hanthorn, J. J., et al. J. Org. Chem. 2012, 77, 6895-6907. [ Links ]
355. Amorati, R., et al. Chem. Eur. J. 2012, 18, 6370-6379. [ Links ]
356. Lynett, P. T., et al. Org. Biomol. Chem. 2011, 9, 3320-3330. [ Links ]
357. Vaidya, V.; Ingold, K. U.; Pratt, D. A. Angew. Chem. Int. Ed. Engl. 2009, 48, 157-160. [ Links ]
358. Zaware, S. B., et al. New J. Chem. 2011, 35, 1615-1623. [ Links ]
359. Litwinienko, G.; Ingold, K. U. J. Org. Chem. 2003, 68, 3433-3438. [ Links ]
360. Litwinienko, G.; Ingold, K. U. J. Org. Chem. 2004, 69, 5888-5896. [ Links ]
361. Litwinienko, G.; Ingold, K. U. J. Org. Chem. 2005, 70, 8982-8990. [ Links ]
362. Litwinienko, G.; Ingold, K. U. Aee. Chem. Res. 2007, 40, 222-230. [ Links ]
363. Foti, M. C. J. Pharm. Pharmaeol. 2007, 59, 1673-1685. [ Links ]
364. Galano, A., et al. Chem. Phys. 2009, 363, 13-23. [ Links ]
365. Urbaniak, A.; Szelag, M.; Molski, M. Comput. Theor. Chem. 2013, 1012, 33-40. [ Links ]
366. Fifen, J. J., et al. Comput. Theor. Chem. 2011, 966, 232-243. [ Links ]
367. Benayahoum, A.; Amira-Guebailia, H.; Houache, O. Comput. Theor. Chem. 2014, 1037, 1-9. [ Links ]
368. Xue, Y., et al. J. Mol. Model. 2013, 19, 3851-3862. [ Links ]
369. Qian, Y. P., et al. Food Chem. 2011, 126, 241-248. [ Links ]
370. Musialik, M., et al. J. Org. Chem. 2009, 74, 2699-2709. [ Links ]
371. Musialik, M.; Litwinienko, G. Org. Lett. 2005, 7, 4951-4954. [ Links ]
372. Dimitric Markovic, J. M., et al. Struet. Chem. 2014, 25, 1795-1804. [ Links ]
373. Najafi, M. Monatsh. Chem. 2014, 145, 291-299. [ Links ]
374. Najafi, M. J. Mex. Chem. Soe. 2014, 58, 36-45. [ Links ]
375. Dorovic, J., et al. J. Mol. Model. 2014, 20, [ Links ]
376. Markovic, Z., et al. Chemieal Papers. 2013, 67, 1453-1461. [ Links ]
377. Van Wenum, E.; Jurczakowski, R.; Litwinienko, G. J. Org. Chem. 2013, 78, 9102-9112. [ Links ]
378. Farmanzadeh, D.; Najafi, M. J. Theor. Comput. Chem. 2013, 12, [ Links ]
379. Bamonti, L., et al. Bioorg. Med. Chem. 2013, 21, 5039-5046. [ Links ]
380. Lengyel, J., et al. PCCP. 2013, 15, 10895-10903. [ Links ]
381. Senthil kumar, K.; Kumaresan, R. Comput. Theor. Chem. 2012, 985, 14-22. [ Links ]
382. Alberto, M. E., et al. Phys. Chem. Chem. Phys. 2013, 15, 46424650. [ Links ]
383. Najafi, M.; Najafi, M.; Najafi, H. J. Theor. Comput. Chem. 2013, 12, [ Links ]
384. Cao, X. Y., et al. Chem. Eur. J. 2012, 18, 5898-5905. [ Links ]
385. Najafi, M.; Zahedi, M.; Klein, E. Comput. Theor. Chem. 2011, 978, 16-28. [ Links ]
386. Najafi, M., et al. Comput. Theor. Chem. 2011, 969, 1-12. [ Links ]
387. Klein, E.; Lukes, V.; Ilcin, M. Chem. Phys. 2007, 336, 51-57. [ Links ]
388. López-Munguía, A., et al. PLoS One. 2011, 6. [ Links ]
389. Cheng, L. X., et al. Org. Biomol. Chem. 2010, 8, 1058-1063. [ Links ]
390. Markovic, Z. S., et al. J. Mol. Model. 2011, 17, 2575-2584. [ Links ]
391. Jeremic, S. R., et al. Monatsh. Chem. 2012, 143, 427-435. [ Links ]
392. Markovic, Z. S., et al. Int. J. Quantum Chem. 2012, 112, 2009-2017. [ Links ]
393. Focsan, A. L.; Pan, S.; Kispert, L. D. J. Phys. Chem. B. 2014, 118, 2331-2339. [ Links ]
394. Markovic, Z., et al. PCCP. 2013, 15, 7370-7378. [ Links ]
395. Nakanishi, I., et al. Org. Biomol. Chem. 2005, 3, 626-629. [ Links ]
396. Ouchi, A., et al. J. Phys. Chem. B. 2009, 113, 13322-13331. [ Links ]
397. Lhiaubet-Vallet, V., et al. J. Phys. Chem. B. 2007, 111, 8277-8282. [ Links ]
398. Estévez, L.; Otero, N.; Mosquera, R. A. J. Phys. Chem. B. 2010, 114, 9706-9712. [ Links ]
399. Marino, T.; Galano, A.; Russo, N. J. Phys. Chem. B. 2014, 118, 10380-10389. [ Links ]
400. Medina, M. E.; Iuga, C.; Alvarez-Idaboy, J. R. PCCP. 2013, 15, 13137-13146. [ Links ]
401. Amorati, R., et al. J. Agrie. Food Chem. 2006, 54, 2932-2937. [ Links ]
402. Bayat, A.; Fattahi, A. Comput. Theor. Chem. 2013, 1018, 35-44. [ Links ]
403. Stepanic, V., et al. Food Chem. 2013, 141, 1562-1570. [ Links ]
404. Amic, D., et al. J. Mol. Model. 2013, 19, 2593-2603. [ Links ]
405. Vagánek, A., et al. Comput. Theor. Chem. 2014, 1050, 31-38. [ Links ]
406. Estévez, L.; Mosquera, R. A. J. Phys. Chem. A. 2008, 112, 10614-10623. [ Links ]
407. Javan, A. J.; Javan, M. J.; Tehrani, Z. A. J. Agrie. Food Chem. 2013, 61, 1534-1541. [ Links ]
408. Leopoldini, M., et al. J. Phys. Chem. A. 2004, 108, 4916-4922. [ Links ]
409. Leopoldini, M.; Russo, N.; Toscano, M. J. Agrie. Food Chem. 2007, 55, 7944-7949. [ Links ]
410. Leopoldini, M., et al. J. Agrie. Food Chem. 2010, 58, 8862-8871. [ Links ]
411. Mazzone, G., et al. Food Chem. 2013, 141, 2017-2024. [ Links ]
412. Mazzone, G., et al. RSC Adv. 2014, 5, 565-575. [ Links ]
413. ;Pérez-González, A., et al. J. Mex. Chem. Soe. 2012, 56, 241-249. [ Links ]
414. Zhang, D., et al. J. Phys. Chem. A. 2013, 117, 1784-1794. [ Links ]
415. Markovic, Z., et al. Food Chem. 2012, 134, 1754-1760. [ Links ]
416. Amic, D.; Lucic, B. Bioorg. Med. Chem. 2010, 18, 28-35. [ Links ]
417. Lespade, L.; Bercion, S. Free Radie. Res. 2012, 46, 346-358. [ Links ]
418. Mohajeri, A.; Asemani, S. S. J. Mol. Struet. 2009, 930, 15-20. [ Links ]
419. Koopmans, T. Physiea. 1934, 1, 104-113.
420. Perdew, J. P., et al. Phys. Rev. Lett. 1982, 49, 1691-1694. [ Links ]
421. Ortiz, J. V. Adv. Quantum Chem. 1999, 35, 33-52. [ Links ]
422. Ortiz, J. V. Wiley Interdiseiplinary Reviews: Computational Mo-leeular Seienee. 2013, 3, 123-142.
423. Galano, A.; Alvarez-Idaboy, J. R. J. Comput. Chem. 2014, 35, 2019-2026. [ Links ]
424. Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B. 2009, 113, 6378-6396. [ Links ]
425. Galano, A.; Tan, D. X.; Reiter, R. J. J. Pineal Res. 2011, 51, 1-16. [ Links ]
426. Corchado, J. C., et al. J. Phys. Chem. A. 1998, 102, 2424-2438. [ Links ]
427. Marcus, R. A. Pure Appl. Chem. 1997, 69, 13-29. [ Links ]
428. Marcus, R. A. Rev. Mod. Phys. 1993, 65, 599-610. [ Links ]
429. Pollak, E.; Pechukas, P. J. Am. Chem. Soe. 1978, 100, 2984-2991. [ Links ]
430. Fernández-Ramos, A., et al. Theor. Chem. Aee. 2007, 118, 813-826. [ Links ]
431. Fernández-Ramos, A., et al. Chem. Rev. 2006, 106, 4518-4584. [ Links ]
432. Kuppermann, A.; Truhlar, D. G. J. Am. Chem. Soe. 1971, 93, 1840-1851. [ Links ]
433. Garrett, B. C.; Truhlar, D. G. J. Phys. Chem. 1979, 83, 2921-2926. [ Links ]
434. Skodje, R. T.; Truhlar, D. G.; Garrett, B. C. J. Phys. Chem. 1981, 85, 3019-3023. [ Links ]
435. Collins, F. C.; Kimball, G. E. J. Colloid Sei. 1949, 4, 425-437. [ Links ]
436. Smoluchowski, M. Z. Phys. Chem. 1917, 92. [ Links ]
437. Einstein, A. Ann. Phys. . 1905, 17, 549-560. [ Links ]
438. Stokes, G. G. Mathematieal and Physieal Papers. Cambridge: Cambridge University Press, 1903. [ Links ]
439. Bielski, B. H. J.; Arudi, R. L.; Sutherland, M. W. J. Biol. Chem. 1983, 258, 4759-4761. [ Links ]
440. Bielski, B. H. J., et al. J. Phys. Chem. Ref. Data. 1985, 14, 1041-1100. [ Links ]
441. Baker, M. Z., et al. Int. J. Radiat Biol. 1982, 41, 595-602. [ Links ]
442. Asada, K.; Kanematsu, S. Agrie. Biol. Chem. 1976, 40, 18911892. [ Links ]
443. Galano, A.; Alvarez-Idaboy, J. R.; Francisco-Márquez, M. J. Phys. Chem. B. 2011, 115, 13101-13109. [ Links ]
444. Joshi, R., et al. J. Agrie. Food Chem. 2005, 53, 2696-2703. [ Links ]
445. Cabelli, D. E.; Bielski, B. H. J. J. Free Radie. Biol. Med. 1986, 2, 71-75. [ Links ]
446. Nadezhdin, A. D.; Dunford, H. B. Can. J. Chem. 1979, 57, 30173022. [ Links ]
447. Nishikimi, M.; Machlin, L. J. Areh. Bioehem. Biophys. 1975, 170, 684-689. [ Links ]
448. León-Carmona, J. R.; Alvarez-Idaboy, J. R.; Galano, A. PCCP. 2012, 14, 12534-12543. [ Links ]
449. Taubert, D., et al. Free Radieal Biology andMedieine. 2003, 35, 1599-1607.
450. Marino, T.; Galano, A.; Russo, N. J. Phys. Chem. B. 2014, 118, 10380-10389. [ Links ]
451. Galano, A.; León-Carmona, J. R.; Alvarez-Idaboy, J. R. J. Phys. Chem. B. 2012, 116, 7129-7137. [ Links ]
452. Mahal, H. S.; Badheka, L. P.; Mukherjee, T. Res. Chem. In-termed. 2001, 27, 595-604. [ Links ]
453. Guha, S. N.; Priyadarsini, K. I. Int. J. Chem. Kinet. 2000, 32, 17-23. [ Links ]
454. Okada, Y., et al. JAOCS, Journal of the Ameriean Oil Chemists' Soeiety. 2010, 87, 1397-1405. [ Links ]
455. Galano, A., et al. Theor. Chem. Aee. 2012, 131, 1-12. [ Links ]
456. Storozhok, N. M., et al. Kinet. Catal. 2004, 45, 488-496. [ Links ]
457. Mitarai, A., et al. J. Agrie. Food Chem. 2008, 56, 84-91. [ Links ]
458. Aruoma, O. I., et al. Free Radie. Res. Commun. 1990, 10, 143-157. [ Links ]
459. Quintiliani, M., et al. Int. J. Radiat Biol. 1977, 32, 195-202. [ Links ]
460. Eriksen, T. E.; Fransson, G. Journal of the Chemieal Soeiety, Perkin Transaetions 2. 1988, 1117-1122. [ Links ]
461. Hofstetter, D.; Nauser, T.; Koppenol, W. H. Chem. Res. Toxieol. 2010, 23, 1596-1600. [ Links ]
462. Kesavan, P. C.; Powers, E. L. Int. J. Radiat Biol. 1985, 48, 223-233. [ Links ]
463. Devasagayam, T. P. A., et al. Bioehimiea et Biophysiea Aeta -Biomembranes. 1996, 1282, 63-70. [ Links ]
464. Brezová, V.; Slebodová, A.; Stasko, A. Food Chem. 2009, 114, 859-868. [ Links ]
465. Poeggeler, B., et al. Redox Report. 1996, 2, 179-184. [ Links ]
466. Matuszak, Z.; Reszka, K. J.; Chignell, C. F. Free Radie. Biol. Med. 1997, 23, 367-372. [ Links ]
467. Stasica, P.; Ulanski, P.; Rosiak, J. M. J. Radioanal. Nuel. Chem. 1998, 232, 107-113. [ Links ]
468. Chyan, Y. J., et al. J. Biol. Chem. 1999, 274, 21937-21942. [ Links ]
469. Marshall, K. A., et al. Free Radie. Biol. Med. 1996, 21, 307-315. [ Links ]
470. Mahal, H. S.; Sharma, H. S.; Mukherjee, T. Free Radie. Biol. Med. 1999, 26, 557-565. [ Links ]
471. Abe, S., et al. Chem. Pharm. Bull. 2004, 52, 186-191. [ Links ]
472. Lin, M., et al. J. Photoehem. Photobiol. B: Biol. 2007, 89, 36-43. [ Links ]
473. Emanuel, C. J., et al. J. Am. Chem. Soe. 1999, 121, 2927-2928. [ Links ]
474. Chatgilialoglu, C., et al. J. Am. Chem. Soe. 2000, 122, 9525-9533. [ Links ]
475. Prior, R. L.; Wu, X.; Schaich, K. J. Agrie. Food Chem. 2005, 53, 4290-4302. [ Links ]
476. Frankel, E. N.; Meyer, A. S. J. Sei. FoodAgrie. 2000, 80, 1925-1941. [ Links ]
477. Antolovich, M., et al. Analyst. 2002, 127, 183-198. [ Links ]
478. Huang, D.; Ou, B.; Prior, R. L. J. Agrie. Food Chem. 2005, 53, 1841-1856. [ Links ]
479. Brand-Williams, W.; Cuvelier, M. E.; Berset, C. LWT - Food Sei. Teehnol. 1995, 28, 25-30. [ Links ]
480. Singleton, V. L.; Orthofer, R.; Lamuela-Raventós, R. M. Methods Enzymol. 1998, 299, 152-178. [ Links ]
481. Benzie, I. F. F.; Strain, J. J. Anal. Bioehem. 1996, 239, 70-76. [ Links ]
482. Cao, G.; Alessio, H. M.; Cutler, R. G. Free Radie. Biol. Med. 1993, 14, 303-311. [ Links ]
483. Huang, D., et al. J. Agrie. Food Chem. 2002, 50, 1815-1821. [ Links ]
484. Miller, N. J., et al. Clin. Sei. 1993, 84, 407-412. [ Links ]
485. Re, R., et al. Free Radie. Biol. Med. 1999, 26, 1231-1237. [ Links ]
486. Wayner, D. D. M., et al. FEBS Lett. 1985, 187, 33-37. [ Links ]
487. Jiménez, A., et al. Org. Lett. 2004, 6, 4583-4586. [ Links ]
488. Porter, W. L. Toxieol. Ind. Health. 1993, 9, 93-122. [ Links ]
489. Frankel, E. N., et al. J. Agrie. Food Chem. 1994, 42, 1054-1059. [ Links ]
490. Frankel, E. N., et al. J. Agrie. Food Chem. 1996, 44, 131-135. [ Links ]
491. De Grey, A. D. N. J. DNA Cell Biol. 2002, 21, 251-257. [ Links ]
492. Terpinc, P.; Abramovic, H. Food Chem. 2010, 121, 366-371. [ Links ]
493. Sies, H. Exp. Physiol. 1997, 82, 291-295. [ Links ]
494. Iuga, C.; Alvarez-Idaboy, J. R.; Vivier-Bunge, A. J. Phys. Chem. B. 2011, 115, 12234-12246. [ Links ]
495. Galano, A.; Francisco-Márquez, M.; Alvarez-Idaboy, J. R. J. Phys. Chem. B. 2011, 115, 8590-8596. [ Links ]
496. Álvarez-Diduk, R.; Galano, A. J. Phys. Chem. B. 2015, 119, 3479-3491. [ Links ]
497. Álvarez-Diduk, R., et al. J. Phys. Chem. B. 2015, 119, 8535-8543. [ Links ]
498. Galano, A.; Francisco-Márquez, M.; Alvarez-Idaboy, J. R. PCCP. 2011, 13, 11199-11205. [ Links ]
499. Galano, A.; Pérez-González, A. Theor. Chem. Aee. 2012, 131, 1-13. [ Links ]
500. León-Carmona, J. R.; Galano, A. J. Phys. Chem. B. 2011, 115, 15430-15438. [ Links ]
501. Galano, A.; Francisco-Márquez, M. J. Phys. Chem. B. 2009, 113, 16077-16081. [ Links ]
502. Pérez-González, A.; Muñoz-Rugeles, L.; Alvarez-Idaboy, J. R. RSC Advanees. 2014, 4, 56128-56131. [ Links ]
1 This principle establishes a linear relation between the reaction enthalpy and the activation energy of a series of closely related chemical processes.

