










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.57 no.4 Ciudad de México oct./dic. 2013
Article
Development and Validation of HPLC Method for Simultaneous Determination of Ceftriaxone and Cefaclor in Commercial Formulations and Biological Samples
Jasmin Shah*, M. Rasul Jan, Sultan Shah, and M. Naeem Khan
Institute of Chemical Sciences, University of Peshawar, Pakistan Fax: 92-91-9216652. jasminshah2001@yahoo.com.
Received March 7, 2013.
Accepted June 3, 2013.
Abstract
A reversed phase high performance liquid chromatographic method has been developed for the simultaneous determination of cefaclor and ceftriaxone cephalosporin antibiotic. The developed method has been validated and applied to mixtures of the commercial formulation and spiked human plasma. A mediterranea C18 column (4.6 x 250 mm) was used with isocratic solvent delivery system and UV-visible detector. Different experimental parameters like solvent composition (acetonitrile: methanol: triethyl amine buffer 1:1:2 (v/v), flow rate of mobile phase (0.6 mLmin-1), pH of the buffer (7), and wavelength (260 nm) were optimized for effective separation and resolution of the analyte peaks. The separation was achieved in 6 min with retention times of 4.94 ± 0.056 min and 3.39 ± 0.022 min for cefaclor and ceftriaxone respectively. The linear range for both the studied drugs was found to be 0.5-250 μgmL-1 with r2 of 0.9987 (cefaclor) and 0.9997 (ceftriaxone). The limit of detection (3.3 σ/S) was found to be 2.34 × 10-2 μgmL-1 and 1.70 × 10-2 μgmL-1, respectively, for cefaclor and ceftriaxone. Similarly limit of quantification (10σ/S) was 7.10 × 10-2 μgmL-1 for cefaclor and 5.15 × 10-2 μgmL-1 for ceftriaxone. The chromatographic procedure was applied to commercial formulations and spiked human plasma and the results were compared with literature HPLC method.
Keywords: cefaclor, ceftriaxone, simultaneous, HPLC, commercial formulation, spiked human plasma.
Resumen
Se desarrolló un procedimiento para la determinación de cefaclor y ceftriaxona, dos antibióticos de la familia de cefalosporinas, por cromatografía de líquidos de alta resolución en fase inversa. El método fue utilizado en la determinación de dos compuestos en mezclas de formulaciones comerciales y en plasma sanguínea enriquecida con ambos. La separación se obtuvo en la columna Mediterranea C18 (4.6 x 250 mm) con elución isocrática. Los parámetros de separación cromatográfica y las condiciones de detección se establecieron con base en los experimentos tomando como criterio la resolución cromatográfica: la composición de fase móvil (acetonitrilo: metanol: tampón de trietilamnia 1:1:2 (v/v), flujo en la columna (0.6 mLmin-1), pH del tampón (pH 7) y la longitud de onda de detección (260 nm). En las condiciones propuestas, la separación de dos compuestos se obtuvo en 6 min con tiempos de retención 4.94±0.056 min y 3.39±0.022 min para cefaclor y para ceftriaxona, respectivamente. El rango lineal para ambos fármacos correspondió al intervalo 0.5-250 μgmL-1 con r2 of 0.9987 (cefaclor) y 0.9997 (ceftriaxona). Los límites de detección (3.3 σ/S) fueron 2.34 × 10-2 μggmL-1 y 1.70 × 10-2 μgmL-1, respectivamente para cefaclor and ceftriaxona. Por su parte, los límites de quantificación (10 σ/S) fueron 7.10 × 10-2 μgmL-1 y 5.15 × 10-2 μgmL-1. El procedimiento desarrollado fue utilizado en la determinación de dos compuestos en formulaciones comerciales y en plasma sanguínea enriquecida, comparando los resultados con los reportados en bibliografía.
Keywords: Cefaclor, ceftriaxona, CLAR, formulaciones comerciales, plasma sanguínea enriquecida con fármacos.
Introduction
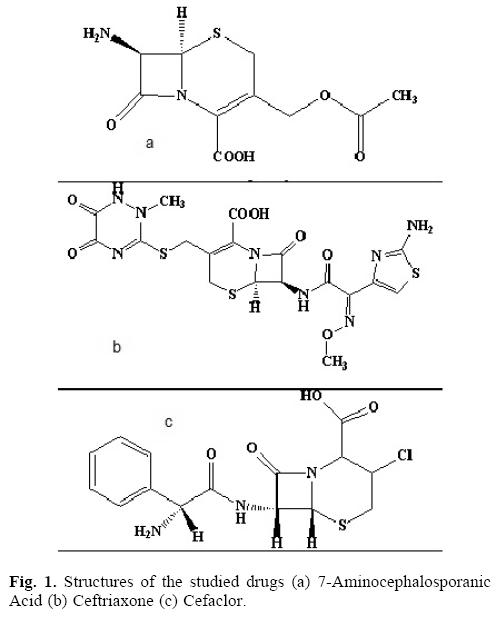
After the isolation of Cephalosporin C from fungus Cephalosporium Acremonium, in the early 1950s and its conversion to 7-aminocephalosporanic acid, a new group called cephalosporin was added to the arsenal of antibiotics. Five generation of this new group are currently in clinical use, all of which are semisynthetic antibacterial agents derived from the 7-ami-nocephalosporanic acid and belong to the β-lactam family of antibiotics. The cephalosporins have greater stability towards deactivation by β-lactamases as compared to other β-lactam antibiotics. Cephalosporins are available in both parenteral and oral form and are among the widely prescribed antibacterial group worldwide [1, 2].
Ceftriaxone is third generation parenteral cephalosporin possessing broad spectrum of activity low MIC90 value (1-4 μgmL-1) for most strains of Enterobacteriaceae and Staphylococcus aureus [3, 4]. Cefaclor is second generation oral cephalosporin antibacterial agent prescribed for the treatment of wide range of pediatric infections including otitis media, tonsillitis and skin infections. However, it causes serum sickness like reaction in children [5].
In the literature a number of analytical methods have been reported for the determination of ceftriaxone and cefaclor in pharmaceutical formulation and biological samples. These include spectrophotometric [6-10], spectrofluorimetric [11-13], and chromatographic [14-24]. In the recent past the quality of drugs especially antibacterial agents has been compromised worldwide particularly in the developing countries. The easy availability of counterfeit/substandard drugs and the misuse of antibiotics have resulted in the development of resistant strains of microbes which is serious challenge for the health professionals. The aim of the present work is to develop a simple, sensitive, cost effect and time saving method for the simultaneous quantification of the two most frequently prescribed cepha-losporins in commercial formulations as well as in biological samples. The simultaneous quantification of the two important cephalosporins with isocratic solvent system in a single run not only saves the solvent but also with a short run time makes it a better choice for the analysis of these drugs in quality control and research laboratories. The proposed method was validated according to ICH guidelines and requires minimum preparation of sample with a well-resolved peaks without interferences.
Experimental
Materials and Reagents
All reagents were of HPLC grade purity or high grade purity. Triethyl amine (TEA), glacial acetic acid, acetonitrile and methanol (BioM laboratories Cerritos, USA) were used in this work. Standard reference ceftriaxone and cefaclor were provided by Cirin Pharmaceutical (Pvt) Ltd., Hattar, Pakistan. Commercial formulations were purchased from local market.
Preparation of Reagent Solutions
Glacial acetic acid (1.0 M) was prepared by diluting 5.7 mL of 17.5 M glacial acetic acid to 100 mL with distilled water. TEA solution (0.1 M) was prepared by diluting 1.4 mL of pure liquid TEA (7.2 M) to 100 mL with distilled water. TEA (0.01 M) buffer solution of pH 7 was prepared by diluting 25 mL of 0.1 M stock solution of TEA and adjusting the pH to 7 with 1 M acetic acid solution. The HPLC grade acetonitrile, methanol and TEA buffer were mixed in 1:1:2 (v/v). The mobile phase was filtered through 0.45 urn nylon membrane filter paper (Millipore).
Preparation of Standard Solution
Standard ceftriaxone solution was prepared by dissolving 0.00625 g of pure drug in water and diluted to 25 mL with distilled water to get stock solution (250 μgmL-1). Similarly, 250 μgmL-1 stock solution of cefaclor was prepared by dissolving 0.00625 g in water on gentle warming and diluted to 25 mL with distilled water. The working standards were prepared by dilution of appropriate volume of stock solutions with mobile phase. The artificial drug mixture, for optimization studies, was prepared by mixing standard stock solution of both the drugs in 1:1 (concentration units).
Instrumentation and Chromatographic Conditions
All the weights were measured with Sartorius GMBH GÜTTINGEN Germany, digital single pan balance (maximum capacity 30.00000g±0.00001 g). The chromatographic system consisted of an Acme 9000 Series HPLC equipped with SP 930 isocratic pump and UV 730 D detector (Young Lin, Korea). All the separations were carried out on C18 column (5μm, 4.6 mm Ø x 250 mm, Teknokroma S. Coop. C. Ltd. Barcelona, Spain) with a guard column containing with identical packing material to that of the analytical column and a rheodyne injector with 20 μL loop were used. Degassing of the mobile phase was carried out with a ks300 KUM SUNG ultrasonic (Korea) sonicator. The biological samples were centrifuged by using CL international clinical centrifuge (USA). A ternary mixture of acetonitrile, methanol and triethyl amine (TEA) buffer (pH 7) in ratio of 1:1:2 v/v was used as mobile phase. The mobile phase was filtered through 0.45 um membrane filter (Milli-pore), degassed by sonication for 30 min and delivered at rate of 0.9 mLmin-1. All the sample solutions were also filtered through micropore filtration assembly (Millipore) before injection. A 20 μl sample was injected to the column by using a 100 μL syringe (Hamilton Co., Reno. Nevada USA) and the column effluents were monitored with UV detector at 240 nm. All measurements were performed at ambient temperature i.e. 25 °C with run time of 10 min.
Construction of Calibration Curve
Artificial mixtures of the standard drugs in the range of 0.5250 μgmL-1 were prepared by mixing appropriate volumes of the standard stock solutions and diluted with mobile phase to the required volume. All standard solutions were filtered through 0.45 μm nylon membrane filter and injected into the column under the optimized chromatographic conditions. The peak height was plotted against the concentration to construct the calibration curve.
Application to Artificial Mixtures of Commercial Formulations
Contents of the five vials labeled to contain 250 mg of the ceftriaxone sodium were weighed and mass of powdered commercial formulation per vial was calculated. Similarly the average weight of powder of commercial formulation per capsule of cefaclor was also calculated. Mass of powdered commercial formulations claimed to contain 0.00625 g of ceftriaxone was dissolved in water and diluted to 25 mL with distilled water to get stock solution (250 μgmL-1). Similarly, 250 μgmL-1 stock solution of cefaclor commercial formulation was prepared by dissolving mass of powdered commercial formulations claimed to contain 0.00625 g of cefaclor in distilled water on gentle heating and diluting to 25 mL with distilled water. Artificial mixture of the commercial formulations (1:1) in the concentration range of 0.5 μgmL-1, 2.5 μgmL-1 and 5.0 μgmL-1 were prepared by mixing appropriate volumes of stock solutions of the commercial formulations and diluting to appropriate volume with the mobile phase. All sample solutions were filtered through 0.45 um membrane filter before injection to the chromatographic system.
Application to Spiked Plasma Samples
Blood samples were provided by Khyber Teaching Hospital, Peshawar and stored at -20 °C. 500 μg standard of each of the drug i.e. ceftriaxone and cefaclor, was added to 5 mL plasma and deproteinized by mixing with 15 mL of acetonitrile. The mixture was centrifuged at the rate of 3500 rpm for 5 minutes. The supernatant was removed and diluted to 50 mL with distilled water. The resulting plasma solution has a concentration of 10 μgmL-1 with respect to both drugs. Plasma samples in concentration range of 0.5 μgmL-1, 2.5 μgmL-1 and 5 μgmL-1 were prepared by dilution with mobile phase. All spiked plasma samples were filtered through 0.45 um membrane filter and injected to the chromatographic system under the optimum chromatographic condition already mentioned.
Results and discussion
Optimization of Experimental Conditions
Ceftriaxone is third generation parenteral while cefaclor is second generation oral cephalosporin and have closely related chemical structures (Fig. 1). A number of experimental conditions were checked for their potential effect on the resolution of chromatographic peaks of the studied drugs. However, four experimental parameters, mobile phase composition, mobile phase flow rate, wavelength and pH were selected for thorough investigation of their effect on the separation of both the drugs.
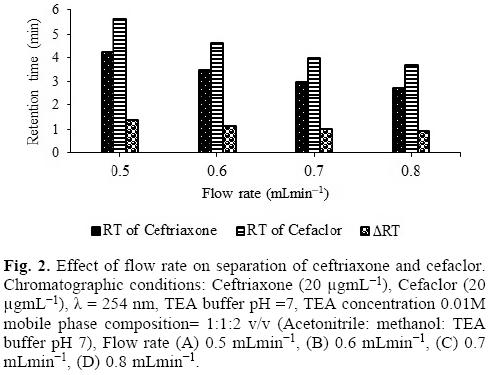
Ternary mixtures of acetonitrile, methanol and TEA buffer in four different compositions (1:0:4, 1:0:3, 1:0:2, 1:1:2) were investigated for their separation using reversed phase HPLC technique. Reasonable separation with symmetrical peaks was obtained with mobile phase containing acetonitrile, methanol and TEA buffer in 1:1:2. The effect of flow rate was investigated from 0.5 mLmin-1 to 0.8 mLmin-1. It was observed that the increase in flow rate decreases the retention time of both the drugs. The retention time of ceftriaxone was 4.94 min and cefixime was 3.39 min at a flow rate of 0.6 mL/min. Keeping in mind the separation of peaks as well as the consumption of solvent, 0.6 mL/min was selected as optimum flow rate (Fig. 2).
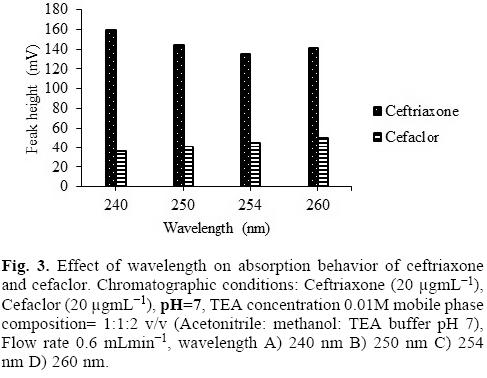
The column eluents were passed through UV detector and monitored at four different wavelengths, 240 nm, 245 nm, 250 nm, 260 nm and the peak heights were evaluated. Maximum peak heights for both the drugs were obtained at 260 nm which was selected as optimum wavelength for rest of the measurements (Fig. 3).
The chromatographic separation of the two drugs was studied at two different pH levels (pH 7 and pH 8). It was found that pH is not affecting the chromatographic separation to greater extent therefore, pH 7 TEA buffer was used in further analysis.
Validation of the method
According to the International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use, guidelines for the validation of analytical procedures, the newly developed method was validated in terms of precision, trueness, specificity, linearity, limit of detection (LOD) and limit of quantification (LOQ).
Linearity and Linear Range
The linear range of the investigated method was studied in the range of 0.5-500 μgmL-1 for both drugs. A linear increase in the peak height was observed with increase in concentration upto 250 μgmL-1 beyond which a positive deviation was noted in both cases.
Limit of Detection and Limit of Quantification
Six replicates at the lowest concentration level of the linear range, for both the studied drugs, were analyzed by the investigated procedure and the standard deviation(s) of the response factor was calculated. The LOD (3.3s/b) and LOQ (10s/b) were then calculated on the basis of standard deviation of response and slope (b). The important analytical characteristics of the proposed method are listed in Table 1.
Precision and accuracy (trueness)
The precision and trueness were evaluated by analysing mixture of the pure standard drugs and mixture of the commercial formulations at three different concentration levels within the linear range by the proposed procedure. Percent recoveries and the relative standard deviations were calculated and Student's t-test was applied to validate the trueness and precision of the proposed method. Similarly, mixture of commercial formulation and spiked human plasma were also analyzed and the percent recoveries and %RSD were calculated. The percent recoveries of ceftriaxone and cefaclor were found to be in the range of 97.68-102.61% (RSD ≤ 4.49) and 98.36103.01% (RSD ≤ 4.85) respectively, in commercial formulations. With spiked human plasma samples, the percent recoveries were in the range of 98.03-103.85% (RSD ≤ 4.92) for both the drugs. The RSD values show good precision of the proposed method for quantification of the studied drugs (Table 2).
The Interday and intraday precision of the proposed method was also determined by analysing binary mixtures of the studied drugs at three different concentration levels by the proposed procedure at different times of the same day and on different days. The low RSD values showed that the proposed procedure exhibit excellent Interday and intraday precision (Table 3).
Selectivity
Chromatograms of the standard drugs in isolated and mixture of standard drugs were compared with those of the commercial formulations and plasma samples for ascertaining the selectivity of the method. The retention time and peak shape remained unchanged in isolated and mixture form showing the selectivity of method for the studied drugs in comparison to the components of the sample matrix (Fig. 4). The other smaller peaks visible in these chromatograms may be due to the impurities or the excipients present in the commercial formulations. Smaller shoulder peaks were also observed at high concentrations and when 4-5 old days standard solution.
The interference effect of matrix of samples was evaluated by using standard addition technique. Mixture of commercial formulations containing known concentration of both the drugs were spiked with known quantities of the standards of both the studied drugs at three different concentration levels were analyzed by the proposed chromatographic procedure (Table 4). The good percent recoveries and low relative standard deviation confirmed that the proposed chromatograph-ic procedure is specific for the studied drugs and free from matrix interferences. The proposed method was also applied for the simultaneous quantification of ceftriaxone and cefa-clor in spiked human plasma (Table 4). The concentration of the studied drug in plasma was selected keeping in mind the quantity that might be present in the blood after four half-lives (approximately 12 hours for cefaclor and 24 hours for ceftriaxone) when a dose of 250 mg of drug is administered to the patient.
Method Application
Samples of two different commercial formulations of both the drugs were simultaneously analysed by the proposed procedure and the quantity of active ingredients were calculated. No HPLC method is available for the simultaneous quantification of both drugs in literature; therefore, two separate reference HPLC methods, one for ceftriaxone [25] and another for cefaclor [26] were used for statistical comparison. The results were compared statistically with the two reference HPLC methods using student's t-test and variance ratio F-test at 95 % confidence level. The calculated t-values and F-values are lower than the theoretical values showing that the proposed method and reference methods are statistically equivalent in terms of precision and trueness. The results obtained from these investigations show that the proposed procedure is suitable for the simultaneous quantification of both the studied drugs in commercial formulation and biological samples.
Conclusion
The running cost, speed of analysis, high degree of specificity and selectivity plays important role in deciding the suitability of an HPLC method for analysis of pharmaceuticals. The use of isocratic HPLC methods for simultaneous quantification of two or more drugs is getting popularity because such methods not only lower the cost but also increase the speed of analysis. The purposed isocratic reverse phase HPLC method is simple, specific and accurate with a single and low cost solvent system for determination of two different cephalosporins. The simultaneous quantification of the two important cephalosporins with isocratic solvent system in a single run not only saves the solvent but also with a short run time makes it a better choice for the analysis of these drugs in quality control and research laboratories.
References
1. Ogorevc, B.; Gomiscek, S. J. Pharm. Biomed. Anal. 1991, 9, 225-236. [ Links ]
2. Eickhoff, T.C.; Ehret, J. Antimicrob. Agents Chemother. 1981, 19, 435-442. [ Links ]
3. Gnann Jr, J.W.; Goetter, W.E.; Elliott, A.M.; Cobbs, C.G. Antimicrob. Agents Chemother. 1982, 22, 1-9. [ Links ]
4. Newton, G.G.; Abraham, E.P. Biochem. J. 1956, 62, 651-658. [ Links ]
5. King, B.A.; Geelhoed, G.C. J. Paediatr. Child Health. 2003, 39, 677-681. [ Links ]
6. El-Walily, A.F.M.; Gazy, A.A.; Belal, S.F.; Khamis, E.F. J. Pharm. Biomed. Anal. 2000, 22, 385-392. [ Links ]
7. Al-Momani, I.F. J. Pharm. Biomed. Anal. 2001, 25, 751-757. [ Links ]
8. Amin, A.S.; Ragab, G.H. Spectrochim. Acta Part A. 2004, 60, 2831-2835. [ Links ]
9. Zhao, W.; Zhang, Y.; Li, Q. Clin Chim Acta. 2008, 391, 80-84. [ Links ]
10. Saleh, G.A.; El-Shaboury, S.R.; Mohamed, F.A.; Rageh, A.H. Spectrochim. Acta Part A. 2009, 73, 946-954. [ Links ]
11. Bebawy, L.I.; El Kelani K.; Fattah L.A. J. Pharm. Biomed. Anal. 2003, 32, 1219-1225. [ Links ]
12. Omar, M.A.; Abdelmageed, Ü.H.; Attia, T.Z. Talanta 2009, 77, 1394-1404. [ Links ]
13. Shah, J.; Jan, M.R.; Shah, S. Naeem, M. J. Fluoresc. 2011, 21, 2155-2163. [ Links ]
14. Gupta, S. Prasad, B.B. J. Pharm. Biomed. Anal. 2000, 23, 307313. [ Links ]
15. Al-Rawithi, S.; Hussein, R.; Raines, D.A.; Al-Showaier, I.; Kurdi, W. J. Pharm. Biomed. Anal. 2000, 22, 281-286. [ Links ]
16. Kai, M.; Kinoshita, H.; Morizono, M. Talanta 2003, 60, 325-334. [ Links ]
17. Chen, X.; Zhonga, D.; Huang, B.; Cui, J. J. Chromatogr. B. 2003, 784, 17-24. [ Links ]
18. Samanidou, V.F.; Hapeshi, E.A.; Papadoyannis, I.N. J. Chromatogr. B. 2003, 788, 147-158. [ Links ]
19. Kai, M.; Kinoshita, H.; Ohta, K.; Hara, S.; Lee, M.K.; Lu, J. J. Pharm. Biomed. Anal. 2003, 30, 1765-1771. [ Links ]
20. Samanidou, V.F.; Ioannou, A.S.; Papadoyannis, I.N. J. Chro-matogr. B. 2004, 809, 175-182. [ Links ]
21. Shrivastava, S.M.; Singh, R.; Tariq, A.; Siddiqui, M.R.; Yadav, J,; Negi, P.S.; Chaudhary, M.A. Int. J. Biomed. Sci. 2009, 5, 37-43. [ Links ]
22. Palanikumar, B.; Thenmozhi, A.; Sridharan, D. Int. J. Pharmacy Pharm. Sci. 2010, 2, 34-36. [ Links ]
23. Sultana, N.; Arayne, M.S.; Shahzad, W. J. Chin. Chem. Soc. 2010, 57, 1278-1285. [ Links ]
24. Nawaz, M.; Arayne, M.S.; Sultana, N. Acta Chromatogr. 2011, 23, 205-213. [ Links ]
25. Belliveau, P.P.; Freeman, C.D.; Nicolau, D.P.; Nightingale, C.H.; Tessier, P.R.; Quintiliani, R. Am. J. Health-Syst. Pharm. 1996, 53, 1024-1027. [ Links ]
26. Signs, S.A.; File, T.M.; Tan, J.S. Antimicrob. Agents Chemother. 1984, 26, 652-655. [ Links ]

