










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.56 no.2 Ciudad de México abr./jun. 2012
Article
Structural and Compositional Optimization of the LiNi0.8Co0.2O2 Electrode by New Synthesis Conditions
Yodalgis Mosqueda Laffita,* Carlos. R. Milian Pila, Mario Pomares Alfonso, Joelis Rodríguez Hernández, and Eduardo Pérez Cappe
Institute of Materials Science and Technology, Havana University, Zapata y G, Plaza de la Revolución, Vedado, 10400, Havana, Cuba. *yodalgis@imre.oc.uh.cu
Received October 10, 2011.
Accepted April 5, 2012.
Abstract
The optimization of Citrate Precursor Method to obtain the LiNi08Co02O2 oxide from the thermal decomposition of the citrate precursor (NH4)3LiNi08Co0.2(C6H5O7) is presented. The optimization procedure consists of both the lithium atmosphere and the reaction time control during the decomposition of the citrate precursor. Were obtained and characterized two kind of the (Li1–xNix)(Ni08Co02)O2 oxides, with and without optimized synthesis conditions, identified as A and B oxides, respectively. The A and B oxides are characterized by compositional, structural and electrochemical studies. The results showed that is possible to reach the ordered oxide phase at smaller reaction time if the lithium atmosphere is controlled. From the combination of the chemical analysis by ICP and the DRX Rietveld structural refinement it is possible to establish the Li, Ni(II), Ni(III) and Co(III) composition with great accuracy. The resulted structural and compositional transformations have a close relation with technological parameters of the rechargeable lithium battery using Li Ni0.8Co0.2O2 oxide as cathode.
Key words: Citrate Precursor, LiNi0.8Co0.2O2, lithium–rechargeable–battery, DRX–Rietveld, ICP–OES analysis.
Resumen
Se presenta la optimización del método citrato para obtener el óxido LiNi0.8Co0.2O2 a partir del precursor (NH4)3LiNi0,8Co0,2(C6H5 O7). La optimización del método consiste en el control de la atmósfera de litio y el tiempo de reacción durante la descomposición del precursor. Fueron obtenidos dos tipos de óxidos de composición general (Li1–xNix)(Ni0.8Co02)O2, con y sin condiciones de optimización, los que fueron denominados A y B, respectivamente. Los óxidos A y B son caracterizados mediante estudios composicionales, estructurales y electroquímicos. Los resultados muestran que es posible obtener la fase ordenada del óxido a menor tiempo de reacción si durante el proceso de síntesis es controlada la atmósfera de Li. Combinando el análisis químico por ICP–OES y la refinación estructural por Rietveld es posible establecer la composición de Li, Ni(II), Ni(III) and Co (III) con exactitud. Se manifiesta una relación muy estrecha entre las transformaciones composicionales y estructurales y los parámetros tecnológicos resultantes para la batería recargable de Li con LiNi0.8Co0.2O2 como cátodo.
Palabras clave: Precursor–citrato, Li– Ni–Co, batería recargable de Li, DRX Rietveld, ICP–OES.
Introduction
The LiNiO2 oxide continues being studied [1–6] owing to their better electrochemical properties in comparison with other systems when they are employed as a cathode material. To avoid the well–known "structural disorder" in 3a positions (called cationic mixture), responsible for a detriment of the electrochemical properties in these compounds, the doping with different transition elements, which partially or totally substitute the nickel ions in its positions (3b), has been one of the used ways [7–29]. Nevertheless of the high cost and toxicity of cobalt as a nickel substitute, the investigations with this transition metal as a doping material continue having a great actuality [7, 9, 10, 12–17, 19, 21, 23, 24, 26, 27, 29–32]. In the last ten years the studies [7, 8, 10, 12, 15, 16, 27] have been directed to diminish as high as possible the cobalt quantity without detriment of the electrochemical behaviour.
It has been reported [8, 13, 15, 17, 32], that ordered LiNi–yCo1–yO2 oxides have a α–NaFeO2 type structure with trigonal symmetry (R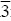 m ) in which the lithium and the trivalent cations (Ni3+ and Co3+) are in the alternating layers of octahedral sites, in the spacial positions 3a (000) and 3b (001/2), respectively. It is known that, when the traditional ceramic method to obtain the nickel rich ( y ≥ 0,6) materials is used, each Li+ substitutes the Ni2+ in the NiO cell, giving rise to a charge unbalance that provokes the oxidation of Ni2+ to Ni3+ for each substituted Ni2+. As the reaction takes place in an air atmosphere, at high temperature, it is always accompanied by loss of lithium. At the same time a quantity of Ni2+ ions is not substituted and not oxidized in the resulted new phase. These impurities of Ni2+ have an ionic radius of (0.63
m ) in which the lithium and the trivalent cations (Ni3+ and Co3+) are in the alternating layers of octahedral sites, in the spacial positions 3a (000) and 3b (001/2), respectively. It is known that, when the traditional ceramic method to obtain the nickel rich ( y ≥ 0,6) materials is used, each Li+ substitutes the Ni2+ in the NiO cell, giving rise to a charge unbalance that provokes the oxidation of Ni2+ to Ni3+ for each substituted Ni2+. As the reaction takes place in an air atmosphere, at high temperature, it is always accompanied by loss of lithium. At the same time a quantity of Ni2+ ions is not substituted and not oxidized in the resulted new phase. These impurities of Ni2+ have an ionic radius of (0.63 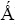 ) and accommodate in the octahedral Li sites (0.74 ), giving rise to a phase mixture of Li+ and Ni2+ excess in the 3a positions. This is usually represented as Li1–x(Ni1–yCoy)1+xO2 (0 ≤ x ≤ 0,6, y ≤0,2) or (Li1–x Nix)(Ni1–yCoy)O2.
) and accommodate in the octahedral Li sites (0.74 ), giving rise to a phase mixture of Li+ and Ni2+ excess in the 3a positions. This is usually represented as Li1–x(Ni1–yCoy)1+xO2 (0 ≤ x ≤ 0,6, y ≤0,2) or (Li1–x Nix)(Ni1–yCoy)O2.
The abundance of Ni2+ over 5% in the Li positions (3a) leads to a change in the cationic arrangement in alternating layers which is observed in the XRD conventional analysis [7, 8, 30–32]. When the occupancy is less than 5% it is necessary to apply most local determinations to observe these irregularities.
Several works have been performed on the structure determination of Li1–xNi1+xO2 and (Li1–x Nix)(Ni1–yCoy)O2 compounds by Rietveld analysis of X–rays data [1, 7, 30–33]. Although most of these works should be considered as being correct, the resolving power of the X–rays method is limited in accordance with the results obtained by Gover et al [30–32]. They show that it is possible to estimate the occupation of 3a position by Ni2+ ions with Rietveld analysis of X–rays data with a difference of 1% in comparison to the results obtained by neutron diffraction data.
It was also noted by Gover et al [30–32] that although the obtained lattice parameters are slightly different when determined by X–rays and neutron diffraction methods, the trends are identical. Therefore, we assume that Rietveld analysis of X–rays data is useful when a routine study to relate the structural disorder and electrochemical properties is needed in samples with different stochiometry and neutron analysis is not accessible.
Independently that the arguments offered by these authors to explain the less resolution of the Rietveld of X–rays data analysis to detect the lighter elements such as Li are valid, some polemic aspects most be mentioned: although lithium ions (Z = 3) have a low scattering factor, the existence of x nickel ions (Z = 28) in 3a sites, is responsible for the XRD patterns sensitivity to lithium deficiency and thus for the accuracy for Rietveld of X–rays data analysis.
On the other hand, studies reveal that the electrochemical properties of the LiNiyCo1––yO2 cathode are extremely dependent of the synthesis conditions [7, 31, 32] and hence its optimization is essential in order to minimize the above mentioned constraints.
Recently we reported [8] a new method of synthesis for nickel rich oxides in which the cationic mixture was minimized and the electrochemical properties improved in the lithium batteries. Nevertheless, the proposed method did not avoid the lithium losses and the time of reaction resulted as high as for the ceramic method (30 h), which prevents to reach the theoretical specific capacity for the lithium batteries application. Published works [7, 31] have shown that there is a sintering time and optimal Li composition for ordering in 3 a sites, which is specific for each oxide composition and the synthesis rout, however few methods reported lithium excess in the initial mixture in order to reduce lithium deficiency and only one [33] showed a systematic study to determine the optimal excess in Lil–xNi1+xO2.
In this work we optimized the synthesis conditions to obtain LiNi08Co02O2 with minimal Ni2+ excess in 3a sites. From the same citrate precursor published in earlier work [8] we demonstrate that the time reaction and Li atmosphere control are necessary to reach a high structural ordering with the higher Li content which allow a best electrochemical behaviour. The different ordering in 3a sites determined by Rietveld X–rays data analysis and ICP–OES technique combination for several samples is compared with the results obtained for samples without this procedure and with those of similar composition reported by other authors. The estimated Li+/Ni2+ occupation in 3a positions has been correlated with changes in the electrochemical charge/discharge properties for different compositions.
Results and discussion
The main focus of this work was centred on the development of a new and more effective method for preparation of LiNi0.8Co0.2O2 oxide that could be used as an efficient positive electrode material for rechargeable lithium batteries. The (Li1–x Nix)(Ni0.8Co0.2)O2 oxides obtained with and without [8] optimized synthesis conditions will be denominated as A and B oxides, respectively. The experimental procedure is showed in Figure 1. This procedure was established by several approximations with the ICP and XRD assistance.
The optimal reaction time to obtain the (Li1–x Nix)(Ni08Co02) ordered oxides (A and B oxides) was established by the XRD study as it is shown in Figure 2. Is notable that from the same citrate precursor [8] the reaction time may be reduced, from 30 h (for the B oxide) to 16 h (for the A oxide) if the lithium atmosphere is controlled. In optimization conditions, for times less than 16 h, the trigonal phase exists, but a clear overlapping of the diffraction lines 006/102 and 108/110 indicated the presence of Ni2+ occupying at least a 5% of the Li ion positions as has been well established by many previous works.
Considering that the high vapour pressure of the Li compounds leads to losses of this element, causing deviations of the desired stoichiometry with a detriment of the gravimetric energy capacity of the cathodic material, we established the conditions of atmosphere control by the assistance of ICP analysis. In table 1 the results of ICP analysis are shown. This allowed us to know the quantity of sublimated Li during citrate precursor decomposition (~2% of dried sample) and at the same time the necessary quantity of Li to be added in excess to have one mol by formula, that in the case of the A oxide resulted to be two times the quantity of Li lost.
In the citrate method, contrary to the traditional ceramic way of synthesis, the ions forming part of the oxide are separated at atomic distances (they form part of the same chemical entity) favouring the diffusion process that takes place in the reorganization of the new atomic arrangement. Moreover, the decomposition of the organic part of the citrate gives rise to a more porous and active material, that favours the reaction with the oxygen of the air. Finally, the control of the Li stoi–chiometry (Li ~ 1 mol by formula) for the A oxide allows the transition metals to be in oxidation state III and the cationic mixture to reach the minimal value. The more important results obtained from the optimization of the reaction time is that the control of lithium atmosphere allows the decreasing in 14 hours of the reaction time established for the all previous method [8] in the searching of the ordered oxide.
Results of the refinement process using the Rietveld method for the A oxide, summarized in table 2 for the optimized synthesis, complement the studies carried out by XRD and ICP on this material.
The obtained Rwp, Rp, χ%2 and the graphic plot from the program Fullprof (see Figure 3) are deemed sufficient to demonstrate the quality of Rietveld study. The crystallographic parameters are in good agreement with those reported by other authors [13, 30–32] in the databases for this kind of lamellar oxides.
The resulted cationic distribution in the A oxide was Li0, 9756Ni2+00192Ni3+0 80Co0 20O2 which is in good agreement with the results obtained by ICP analysis.
Figure 4 shows the first ten cycles of deinsertion (charge) and insertion (discharge) carried out in a electrochemical lithium battery with A and B oxides as cathodes at constant current of 30 μA . For comparison is inserted (picture c) the electrochemical behaviour to the first cycle of (Li1–x Nix)(Ni08Co0.2)O2 obtained by ceramic traditional method [8]. The existence of such a small quantity of Ni2+ in the Li+ sites (2%) has no relevant effect over the electrochemical behaviour for A oxide contrary to that observed in B and more drastically in ceramic oxides where the Ni2+ content is higher than 5%.
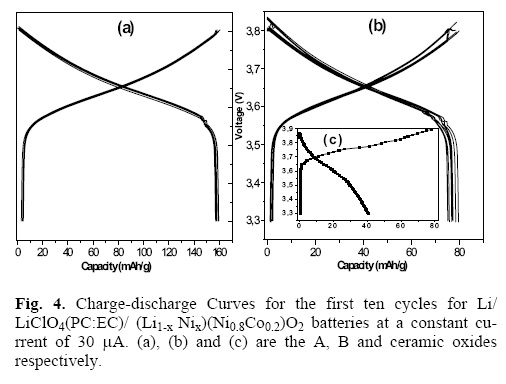
The monotonous increment of the voltage curves against composition for A oxide suggests the occurrence of the chargedischarge monophasic reaction, indicating that the Li deinsertion and insertion in the studied composition interval takes place in all the bulk of the sample, with a great reversibility from one cycle to another. This fact is a consequence of the cationic order in 3a sites and the resulted particle size in the nanoscale dimension obtained, when the solid state precursor method in rich lithium atmosphere is used.
The effect of the enhancement of the Li+ / Ni2+ content in 3a site over the electrochemical behaviour is more notorious in the lithium battery capacity and coulombic efficiency (capacity retention) as it is shown in Figure 5.
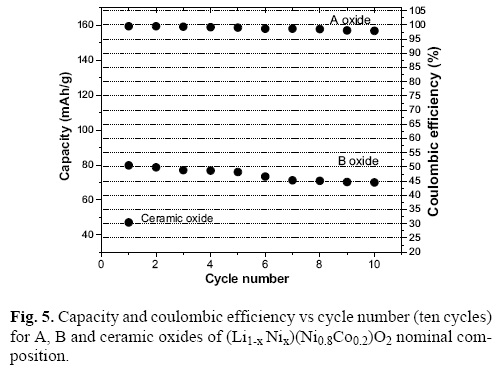
The A oxide shows the double of capacity than B oxide. In addition the A oxide almost reaches the 100% recovery of coulombic efficiency sustained during the first ten cycles which is in relationship with de major Li+ (98%) and minor Ni2+ (2%) content in 3a sites. The ceramic oxide having more than 5% of Ni2+ in 3a sites suffers a fast electrochemical deterioration.
The above behaviour is consistent with previous reports of other authors [7, 31, 32] who have obtained a reduction of capacity with the decreasing of Li in 3a sites. These results suggest that there is an optimum time reaction and lithium composition required for a good structural ordering and electrochemical behaviour for the Li–Ni–Co oxides obtained by citrate method, which in this case resulted to be of 16 hours and 4% Li excess.
Conclusion
It was possible to optimize the conditions of the citrate method of synthesis for the cathodic material LiNi0,8Co0,2O2 The thermal decomposition of the precursor (NH4)3LiNi0,8Co0,2(C6H5O7)2, at 750 °C during 16 h in lithium excess controlled atmosphere, guaranteed the cationic desirable Li composition and the necessary ordering expected to use the potentialities of this oxide as a cathodic material in rechargeable Li batteries. The refinement of Li+ /Ni2+ occupation in the 3a positions by XDR Rietveld, allowed to estimate the composition as Li0,98Ni0,8Co0.2O2. The transformations obtained are reflected in the improved technological parameters as the capacity and coulombic efficiency.
Experimental
Synthesis
The optimization of the reaction time in the synthesis of the LiNi0,8Co0,2O2 oxide, from the thermal decomposition of mixed citrates of Li, Ni and Co (previously synthesized [8] ) at 750 °C during 10, 16 , 20, 25 and 30 h, was carried out. During the optimal time was selected, an experiment assisted by ICP to control the Li content in the oxide was designed. The procedure is represented in Figure 1.
The (Li1–x Nix)(Ni0.8Co0.2)O2 oxides obtained with and without above optimized synthesis conditions will be denominated as A and B oxide, respectively.
Characterization
The quantitative characterization of the three major elements Li, Ni and Co was carried out in an ICP spectrometer model 3300 DV (Perking–Elmer, USA) in the axial mode of observation. The analytical procedure was recently published [34] by us for these materials.
The powder patterns were obtained in a PHILIPS equipment (PW 1710 model) using Cu–Kα1 (1.5406  ) radiation at 42 kV, from 10° to 120° in two theta, with scanning speed of 0.01°/s for the refinement and 2°/s for the phase identification. Five parameters were refined by the Rietveld Method using a FullProf program. The Pseudo–Voigt function was used to fit the peak's profile. The proportions referred to the Li+ and the Ni2+ contents for the 3a position were refined through the occupation factor. Additionally proportion referred to the Ni3+ and the Co3+ contents for the 3b position were fixed in 4:1 ratio.
) radiation at 42 kV, from 10° to 120° in two theta, with scanning speed of 0.01°/s for the refinement and 2°/s for the phase identification. Five parameters were refined by the Rietveld Method using a FullProf program. The Pseudo–Voigt function was used to fit the peak's profile. The proportions referred to the Li+ and the Ni2+ contents for the 3a position were refined through the occupation factor. Additionally proportion referred to the Ni3+ and the Co3+ contents for the 3b position were fixed in 4:1 ratio.
The electrochemical measurements were carried out in a Li/LiClO4(PC:EC)/ (Li1–x Nix)(Ni08Co02)O2 cell. The positive electrode consisted of a mixture of oxide and carbon black sintered pellets (0.13 cm diameter) pressed at 3T/cm2. The cells, assembled in an argon–filled dry box, were galvanostatically cycled under 30 μA current values.
Acknowledgements
Financial support from the Grant 07–149 of The Academy of Science for Developing Word, The Alma Mater–2011 National Projects as well as the Project CONACYT/148997 is gratefully acknowledged.
References
1. Kim, C. J.; Ahn, I. S.; Cho, K. K.; Lee, S. G.; Chung, J. K. J. Alloys. Compd. 2008, 449, 335–338. [ Links ]
2. Laubach, S.; Laubach, S.; Schmidt, P. C.; Ensling, D.; Schmid, S.; Jaegermann, W.; Thißen, A.; Nikolowski, K.; Ehrenberg, H. Phys. Chem. Chem. Phys. 2009, 11, 3278–3289. [ Links ]
3. Coung–Ah, J.; Sung–Kwan, K.; Cang–Soo, K.; Dae–Hee, W.; Byung–Il, K.; Kwang–Soo, N. J. Electroceram. 2006, 17, 674671. [ Links ]
4. Han, C. H.; Kim, J. H.; Paeng, S. H.; Kwak, D. J.; Sung, C. M. Thin Solid Films 2009, 4215–4217. [ Links ]
5. Cao, J.; Guo, C.; Zou, H. J. Solid State Chem. 2009, 182, 555–559. [ Links ]
6. Fergus, J. W. J. Power Sources 2010, 195, 939–954. [ Links ]
7. Jang, W. L.; Lu, C. M.; Hwang, W. S.; Chen, W. C. J. Eur. Ceram. Soc. 2010, 30, 503–508. [ Links ]
8. Hwang, B. J.; Santhanam, R.; Chen, C. H. J. Power Sources 2003,114, 244–252. [ Links ]
9. Mosqueda, C.; Pérez–Cappe, E.; Ruiz–Hitzky, E.; Aranda, P. Eur. J. Inorg. Chem. 2005, 13, 2698–2705. [ Links ]
10. Park, M. S.; Hyun, S. H.; Nam S. C.; Cho S. B. Electrochim. Acta 2008, 53, 5523–5527. [ Links ]
11. Zhu , X. J.; Liu, H. X.; Gan, X. C.; Cao, M. H.; Zhou, J. W.; Chen, X. Q.; Ouyang, S. X. J. Electroceram. 2006, 17, 645–649. [ Links ]
12. Khomane, R. B.; Agrawal, A. C.; Kulkarni, B. D.; Gopukumar, S.; Sivashanmugam, A. Mater. Res. Bull. 2008, 43, 2497–2503. [ Links ]
13. Tong, D. G.; Tang, A. D.; Chu, W.; Tang, L. X.; Huang, K. L.; He, C.; Ji, X. C. Mater. Chem. Phys. 2008,107, 385–391. [ Links ]
14. Li, D.; Peng, Z.; Ren, H.; Guo, W.; Zhou, C. Mater. Chem. Phys. 2008, 107, 171–176. [ Links ]
15. Huang, J.; Cang, J.; Li, W.; Cai, W.; Jiang, Z. Thin Solid Films 2008, 516, 3314–3319. [ Links ]
16. Xiao, L.; Cang, C.; Zhao, C.; Ai, X.; Cang, H.; Cao, C. Electrochim. Acta. 2008, 53, 3007–3012. [ Links ]
17. Dahbi, M.; Saadoune, I.; Amarilla, J. M. Electrochim. Acta. 2008, 53, 5266–5271. [ Links ]
18. Song, M. K.; Hong, S. D.; No, K. T. J. Phys. Chem. Solid. 2008, 69, 1249–1252. [ Links ]
19. Sathiyamoorthi, R.; Chandrasekaran, R.; Gopalan, A.; Vasudevan, T. Mater. Res. Bull. 2008, 43, 1401–1411. [ Links ]
20. Ma, X.; Wang, C.; Han, X.; Sun, J.; Sun, J. J. Alloys. Compd. 2008, 453, 352–355. [ Links ]
21. Kim, B. H.; Kim, J. H.; Ha, S. J.; Hong, S. J.; Song, M. C. J. Electroceram. 2009, 23, 447–451. [ Links ]
22. Zhu, Ch.; Cang, Ch.; Cang, W. D.; Hsieh, Ch. C.; Csai, H. M.; Chen, C. S. J. Alloys Compd. 2010, 496, 703–709. [ Links ]
23. Saadoune, I.; Labrini, M.; Cahya, M.; Almaggoussi, A.; Ehrenberg, H. Electrochim. Acta. 2010, 55, 5180–5185. [ Links ]
24. Mukai, K.; Sugiyama, J.; Aoki, C. J. Solid State Chem. 2010, 183, 1726–1732. [ Links ]
25. Cheralathan, K. K.; Kang, N. C.; Park, H. S.; Lee, C. J.; Choi, W. Ch.; Ko, C. S.; Park, C. K. J. Power Sources 2010, 195, 1486–1494. [ Links ]
26. Song, M. C.; Kwon, I.; Shim, S.; Song, J. H. Ceram. Int. 2010, 36, 1225–1231. [ Links ]
27. Zeng, X. L.; Wu, P.; Luo, F. L.; Zhou, C.; Tong, D. G. J. Phys. Chem. Solids 2010, 71, 1404–1409. [ Links ]
28. Bentaleb, C.; Saadoune, I.; Maher, K.; Saadi, L.; Fujimoto, K.; Ito, S. J. Power Sources 2010, 195, 1510–1515. [ Links ]
29. Hua–jun, G.; Xin–hai, L.; Jie, X.; Zhi–xing, W.; Wen–jie, Peng.; Qian–ming, S. Ener. Convers. Manage 2010, 51, 247–252. [ Links ]
30. Gover, R. K. B.; Conemura, M.; Hirano, A.; Kanno, R.; Kawamoto, C.; Murphy, C.; Mitchell, B. J.; Richarson, J. W. J. Power Sources 1999, 81–82, 535–541. [ Links ]
31. Gover, R. K. B.; Kanno, R.; Mitchell, B.; Hirano, A.; Kawamoto, C. J. Power Sources 2000, 90, 82–88. [ Links ]
32. Gover, R. K. B.; Kanno, R.; Mitchell, B.; Conemura, M.; Kawamoto, C. J. Electrochem. Soc. 2000, 147, 4045–4051. [ Links ]
33. Kanno, R.; Kubo, H.; Kwamato, C.; Kamiyama, T.; Izumi, F.; Takeda, C.; Takano, M. J. Solid State Chem. 1994, 110, 216–225. [ Links ]
34. Mosqueda, C.; Pomares, M.; Pérez–Cappe, E.; Fariñas, J. C.; Miranda, A. M.; Larrea, T. Anal. Bioanal. Chem. 2006, 386, 1855–1862. [ Links ]

