










Serviços Personalizados
Journal

Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista mexicana de ingeniería química
versão impressa ISSN 1665-2738
Rev. Mex. Ing. Quím vol.11 no.1 Ciudad de México Abr. 2012
Catálisis, cinética y reactores
Industrial alumina as a support of MoP: catalytic activity in the hydrodesulfurization of dibenzothiophene
Alúmina industrial como soporte de MoP: actividad catalítica en la hidrodesulfuración de dibenzotiofeno
A. Montesinos-Castellanos1, E. Lima2, A. Vázquez-Zavala3, J. A. de los Reyes 3* and M. A. Vera3
1 Departamento de Ingeniería Química, Tecnológico de Monterrey, Av. Eugenio (Garza- Sada 2501 Sur, Col. Tecnológico C.P. 64849, Monterrey, N.L., México
2 lnstituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito exterior s/n, Cd. Universitaria, Del. Coyoacán, CP 04510, México D.F., México.
3 División Ciencias Básicas e Ingeniería, Universidad Autónoma Metropolitana- Iztapalapa, Av. San Rafael Atlixco 186, Col. Vicentina, Iztapalapa, México D.F., 09340.*Corresponding author. E-mail: jarh@xanum.uam.mx
Received 5 of April 2011
Accepted 31 of October 2011
Abstract
Alumina support with a high surface area was used to prepare alumina-supported MoP catalysts, varying the Mo and P loadings from 4% wt. to 53%wt. Mo and P were loaded with an atomic ratio of Mo:P=1. The presence of different phosphate species was detected on all calalysts by NMR and electron diffraction techniques. The A1PO4 phase was identified for highly loaded MoP catalysts (> 26 wt% of theoretical MoP). The catalytic materials were tested in hydrodesulfurization (HDS) of Dibenzothiophene (DBT) under high temperature (5935 K )and pressure (5.5 MPa ) conditions and the highest activity for HDS of DBT was obtained with the catalyst containing Mo 9.9 wt% (13% wt of theoretical MoP) keeping an atomic ratio P/Mo=l. A structural model is proposed considering the Mo and P loading. The catalyst with the best activity showed high surface area (>200 m2/g).
Keywords: alumina, hydrodesulfurization, dibenzothiophene, phosphide, phosphate.
Resumen
Alúmina de alta área superficial se utilizó para preparar catalizadores de MoP soportado variando la cantidad de Mo y P adicionado al catalizador con una relación molar constante de Mo:P=l. La presencia de diferentes especies de fosfatos se puso en evidencia en los materiales catalíticos mediante técnicas de resonancia magnética nuclear y difracción de electrones. La fase A1PO4 se identificó principalmente en los catalizadores con alto contenido de MoP (>26% en peso» de MoP teorico). Los materiales se evaluaron mediante la reacción de hidrodesulfuración de dibenzotiofeno en condiciones de alta temperatura (593 K ) y presión (5.5 MPa ) encontrándose que la mejor actividad fue para el catalizador con un contenido de 9.9% en peso de Mo (13 % en peso de MoP teorico). Como resultado de estas observaciones proponemos un modelo estructural considerando la cantidad de Mo y P agregado al catalizador. El catalizador con mayor actividad tuvo alta área superficial (>200 m2/g) y las partículas más pequeñas de la fase MoP. Estos hechos marcan el potencial que tiene el MoP para aplicaciones a escala industrial.
Palabras clave: alúmina, hidrodesulfuración, dibenzotiofeno, fosfuro, fosfato.
1 Introduction
Excess SOx emissions resulting from diesel combustion have been the cause of more stringent environmental regulations. The most common refinery process for reducing sulfur level is hydrodesulfurization, HDS (Song, 2003; Babich et al., 2003). Alumina-supported Mo or W promoted by Ni or Co are commonly used as catalysts in HDS processes. Sulfur levels, however, do not reach concentrations as low as 15 ppm. Then there is a need to develop new, more active catalysts.
Transition metal nitrides (Sajkowski and Ojama, 1996; Aegerter et al., 1996), carbides (McCrea et al.,1997; Furimsky, 2003) and phosphides, (Oyama et al., 2001; Clark and Oyama, 2003; Clark et al., 2002) have shown outstanding characteristics for hydrodesulfurization and related reactions such as hydrodenitrogenation (HDN). Phosphides, however, are more stable than nitrides and carbides (Nagai et al., 2000; Phillips et al., 2002).
Some transition metal phosphides have shown high HDS and HDN activities, with MoP and Ni2P being among the most active materials. While most studies have been focused on Ni2P (Stinner et al., 2001; Stinner et al., 2002), a relatively small number have been devoted to MoP materials (Oyama, 2003; Wu et al., 2004). Stinner et al. (2000) observed that bulk MoP was six times more active than bulk MoS2 in the HDN of orto-propilaniline. It has been shown that HDS in a model liquid mixture with DBT, quinoline, benzofuran, tetralin and tetradecane proceeds faster using a catalyst 13 % wt MoP/Al2O3 than using a NiMoS/Al2O3 catalyst. Besides, Phillips et al. (2002) have shown that a supported MoP catalyst exhibited an HDS activity four times higher than that of a supported MoS2.
Molybdenum phosphide is usually synthesized by Temperature-Programmed Reduction at high temperatures (823- 1123 K) under a hydrogen flow (Zuzaniuk and Prins, 2003). This high temperature treatment is required to reduce strong P-O bonds, especially in supported materials where interactions between the phosphorous-containing compound and the support occur (Yang et al., 2006). Unfortunately, high temperature treatments lead to large supported particles and sometimes induce a loss of surface. Phosphide catalysts have been synthesized on carriers exhibiting surface areas in the range 90-200 m2.g-1 before impregnation or reduction of the impregnated precursor (Oyama et al., 2001; Phillips et al., 2002). Nevertheless, these high temperature treatments induced a decrease of surface area values (up to ca. 50 %), which is not suitable for an industrial catalyst. HDS industrial catalyst carriers should exhibit a high surface area (>200m2/g) and be produced at a low cost. SiO2 support is a low-cost material but with low interaction with the active phases; this could produce high particle sizes of active phases. Nonetheless, y-Al2O3 is the most widely used support for HDS catalysts due to its outstanding textural and mechanical properties and its relatively low cost. Several studies have shown that it is possible to synthesize highly stable Al2O3 that offers high surface area (Euzen et al., 2002). If surface area does not vary drastically after high-temperature treatments, a phosphide catalyst with a more convenient texture can be obtained. For instance, some of us (Montesinos-Castellanos et al., 2008) have shown that a high surface stable alumina support is able to support MoP particles with different volume/surface ratios.
A few recent publications (Montesinos-Castellanos et al., 2007; Montesinos-Castellanos et al., 2007b) have reported more extensively on the structural properties of these solids, displaying a schematic model for MoP supported on alumina synthesized by the reduction of impregnated materials. Particle sizes of Mo and P species were correlated with both the P/Mo atomic ratio and the reduction temperature.
Regarding the AlPO4 phase formation, Iwamoto and Grimblot (1999) reported that these species could decrease the Mo-support interaction which leads to larger MoS2 crystallites. AlPO4 phase has been observed when P/Mo was higher than 3 in MoP/Al2O3 catalysts.
In order to find the optimal loading of Mo and P on a high surface alumina to reach a balance in active species, MoP catalysts supported on industrial alumina with different Mo and P loadings were investigated using a constant P to Mo atomic ratio (P/Mo=1). These catalysts were characterized by XRD, HRTEM, 27Al and 31P MAS-NMR. An industrial alumina was used as a support in order to consider a carrier with relevant textural properties for the HDS conditions.
2 Experimental
2.1 Catalysts
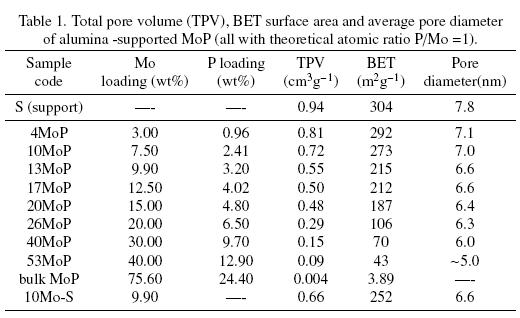
An industrial y-alumina (S), provided by Instituto Mexicano del Petróleo, was used as a support. The surface area of alumina was 360 m2g-1. MoP/Al2O3 catalysts were prepared by incipient wetness impregnation of the support with aqueous solutions of (NH4)6Mo7O24 4H2O and (NH4)2HPO4. The Mo loading was varied from 3 to 40 wt% and the P loading was tuned to maintain the theoretical atomic ratio P/Mo=1; these catalysts have been designated as 4MoP, 10MoP, 13MoP, 17MoP, 20MoP, 26MoP, 40MoP and 53MoP corresponding to different metalP loadings as MoP phase, for instance 13MoP means a catalysts with ~ 13 wt% of MoP phase (Table 1). The materials were dried at 393 K and calcined at 823 K under an air flow; these materials were called precursors. After calcination, the materials were reduced at 1123 K for 2 h under a hydrogen flow (1500 NTP cm3/g min),then cooled down to room temperature under a He flow and sometimes some samples were passivated under a flow of 0.5% O2/He for 2 h.
Two reference materials were prepared: an unsupported MoP (bulk catalyst) sample was synthesized using aqueous solutions of (NH4)6Mo7O24-4H2O and (NH4)2HPO4 with adequate quantities to keep the theoretical atomic ratio P/Mo=1 and a MoS2/Al2O3 sample. In a typical preparation, the support was impregnated with an aqueous solution of (NH4)6Mo7O24 4H2O, then dried under air at 393 K and calcined at 773 K for 4 h . The resulting oxide phase was sulfided at 673 K for 1 h with a mixture of H2S/H2 (15% H2S) at a total flow rate of 66.66 NTP cm3g-1min-1 and a heating rate of 10 Kmin-1, finally the catalyst was cooled to room temperature and flushed with N2 for about 30 min and kept in sealed bottles under argon; this sample was designated as 10Mo-S.
2.2 Characterization
2.2.1 N2 adsorption measurements
Nitrogen adsorption-desorption isotherms were obtained on an Autosorb Quantachrome apparatus. Prior to nitrogen adsorption, the samples were outgassed at 473 K for 4 h. Pore size distributions were calculated from desorption volume plots using the BJH method. The specific surface areas were determined by the BET equation.
2.2.2. Temperature-Programmed Reduction
Temperature-Programmed Reduction (TPR) profiles were obtained on an ISRI RIG 100 apparatus equipped with a thermal conductivity detector and the reducing agent was 10 vol% H2/Ar. The TPR profiles were measured from 373 to 1223 K at 10 K/min. Prior to the analyses, the precursors were dried in the TPR apparatus under an air flow at 393 K for 4 h.
2.2.3. X-ray diffraction
X-ray diffractograms were obtained on a Siemens D500 diffractometer with a copper anode tube. The Kα radiation was selected with a diffracted beam monochromator. Crystal size was calculated using the Scherrer equation D = Kλ/β cos θ, where K = 0.9.
2.2.4. Nuclear Magnetic Resonance
The solid state one pulse nuclear magnetic resonance (NMR) spectra were acquired under magic angle spinning (MAS) conditions using a Bruker Avance II spectrometer with a magnetic field strength of 7.05 Tesla, corresponding to a 31P Larmor frequency of 121.4 MHz. The spectra were acquired with a sample spinning rate of 5 kHz. The chemical shifts were referenced to a H3PO4 solution (85%).
27Al MAS NMR spectra were acquired by operating the spectrometer at 78.3 MHz. Small 27Al flip angles were used (15°) to ensure uniform excitation of all spins. A recycle time of 0.5 s was used. The 27Al chemical shifts were referenced to aqueous [Al(H2O)6]3+.
In order to collect the NMR data on the reduced samples, they were placed in a glovebox under argon and were packed in ZrO2 rotors immediately after preparation. The time taken to record one NMR spectrum was no longer than 10 min.
2.2.5. Conventional and High Resolution TEM
TEM images of several catalysts were obtained using two electronic microscopes. Conventional transmission electron microscopy (TEM) images were obtained using a JEOL-100CX electron microscope with a point- to-point resolution of 0.35 nm. High resolution images were obtained using a JEOL 2010 Field Emission Electron Microscope equipped with an ultra-high-resolution pole piece (λ = 0.00251 mm, resolution of 0.17 nm). The appropriate software (GATAN) was used to obtain interplanar distances and particle size.
2.3 Catalytic tests
2.3.1 Activity
The dibenzothiophene HDS reaction was performed using reagents purchased from Sigma-Aldrich. A batch reactor (250 cm3), stirred slurry Parr 4842, operating at 593 K and 5.5 MPa, was used. The model liquid mixture containing DBT (purity of 99%) at 670 ppm of S and hexadecane (99% purity) was placed in the reactor with 0.2 g of the catalyst. The catalyst was used without a passivation step and was placed in an Ar controlling atmosphere. The reaction was carried out for 8 h and the composition was monitored by manual sampling. The evolution of the reaction was followed by chromatography in a gas chromatograph equipped with a flame ionized detector and a capillary column of 5% phenyl-95% methylpolysiloxane (EC5), purchased from Alltech. First-order rate constants were calculated via a pseudo first-order equation and the integral method (Laredo et al., 2001).
The compounds produced during the reaction were biphenyl, bicyclohexyl and tetrahydrodibenzothiophene, with biphenyl as ca. 90% of the HDS products.
The optimal conditions were found to avoid reaction control by either intraparticle or interfacial diffusion.
2.3.2 Kinetic model
The catalytic activity was considered in terms of the rate constant for the HDS of DBT reaction, assuming a pseudo first-order reaction referred to DBT. This is a reasonable assumption since hydrogen is as a large excess and the reaction conditions allow one to neglect the adsorption terms for H2 and H2S in the expression of the rate of reaction. The constant rate was estimated from a mass balance on DBT, whose integrated form is shown in equation (1).
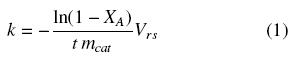
where k is the constant rate (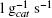 ) calculated by a least-squares fitting, mcat is the catalysts weight (gcat), t is the time (s), Vrs is the mixture volume (l) and XA is the conversion of DBT. In all the cases, correlation coefficients were higher than 0.98. The rate constant in a surface area basis, k'(l m-2 s-1) was obtained by multiplying k by the SBET of the sample.
) calculated by a least-squares fitting, mcat is the catalysts weight (gcat), t is the time (s), Vrs is the mixture volume (l) and XA is the conversion of DBT. In all the cases, correlation coefficients were higher than 0.98. The rate constant in a surface area basis, k'(l m-2 s-1) was obtained by multiplying k by the SBET of the sample.
3 Results and discussion
3.1 Structural aspects
In Fig. 1 the XRD patterns of three selected supported MoP catalysts are compared to those for the support and the unsupported MoP sample (bulk MoP). No diffraction peaks were observed for the carrier and the base line was flat because the alumina was badly crystallized. Samples with low and medium load exhibited XRD patterns similar to the support. On the contrary, for samples with high loading (53MoP), as well as the unsupported MoP sample, peaks at 28, 32, 43, 57.5, 58, 65, 67 and 68 degrees (29) were clearly resolved, corresponding to planes (001), (100), (101), (110), (002), (111), (200) and (102) of the MoP phase, respectively. The peak intensities increased with the Mo and P content. These results suggest that no crystalline phases are present for samples with low and medium loading. These samples, however, were reduced under similar conditions to 53MoP and bulk samples; if MoP is present, particles smaller than 3 nm should be formed. This dimension exceeds the XRD detection limits.
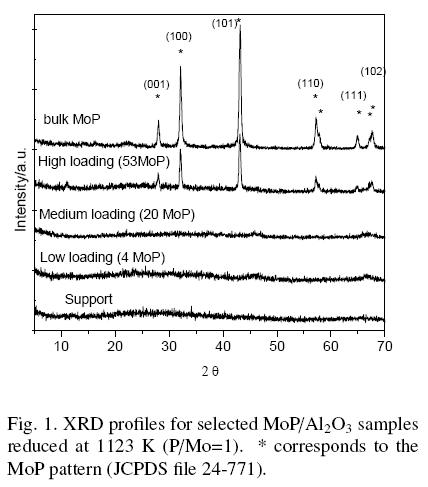
For the highly phosphorous-loaded catalysts, samples 40MoP and 53MoP, the phase AlPO4 was not detected, within the XRD detection limits. Thus, it can be assumed that if any AlPO4 was formed, it is present as an amorphous phase or as microcrystallites (< 3 nm).
Table 2 reports the average crystal size for unsupported MoP and 53MoP samples as determined by the Scherrer equation. The crystal size for 53MoP supported samples was larger than for the unsupported sample (bulk MoP), suggesting a sintering of particles in the reduction step. The bulk MoP sample was reduced at 923 K and the supported samples at 1123 K, causing an increase in crystallite size.
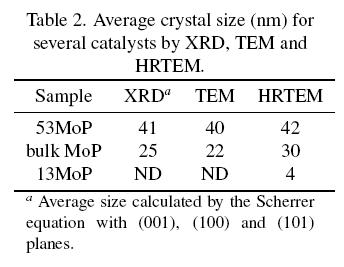
31P MAS-NMR spectra for samples studied in this work exhibited good signal/noise ratio and the NMR peaks were well resolved. In order to find the position of the various isotropic peaks for different phosphorous species, the spectrum for a selected supported reduced then passivated sample (40MoP) were first obtained at different spinning rates. Fig. 2 shows that one isotropic peak appeared at 214 ppm, attributed to a phosphide species. Other peaks at a stronger field (-7 and -14 ppm), due to phosphate groups, were also identified. All other peaks corresponded to spinning side bands for the phosphide NMR signal.
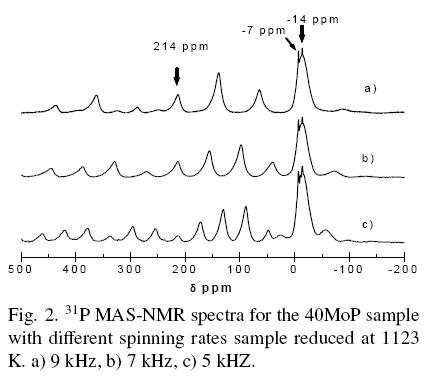
Fig. 3 shows the 31P-MAS NMR spectra for the bulk MoP reduced sample and its corresponding precursor. The spectrum for the reduced bulk MoP showed two isotropic peaks, at ~213 ppm and--2 ppm. The first one corresponded to molybdenum phosphide and the second one was attributed to phosphate species. High anisotropy on the phosphide NMR signal revealed that phosphorus resides on sites that look different when the site is inspected from different directions, which coincides with other studies (Phillips et al., 2002; Zuzaniuk and Prins, 2003). The 31P-MAS NMR spectrum for the precursor of the bulk MoP sample (before the reduction step) showed a single peak close to ~ -2 ppm and a shoulder at -9 ppm corresponding to phosphate and polyphosphate species, respectively. Oyama et al. (2001) reported that MoP precursors have an estimated formula close to oxide MoPO5.5 where phosphorus has a high oxidation state as in phosphates. The precursor sample presented a dark blue color related to molybdenum phosphate glasses. The blue color resulted from intervalence charge-transfer transitions between the Mo5+ and Mo6+ centers (Stinner et al., 2000). Bridge and Patel (1986) proposed that molybdenum phosphate is an interlinked network of near octahedral MoO6 and tetrahedral PO4. The NMR spectrum for the precursor spectrum supports the presence of mono and polymeric molybdenum phosphates. The peak at -2 ppm of the spectrum of the reduced bulk MoP sample was attributed to phosphates. This suggested that the reduction reaction step was probably a shell-to-core process. 31P-MAS NMR spectra for the bulk MoP sample after passivation processes (spectrum not shown) presented significant differences in relation to the bulk MoP reduced sample; the MoP in the bulk sample was very stable under air.
The 31P MAS NMR spectra for selected MoP supported samples after passivation processes, Fig. 4, presented the characteristic peaks of mono and polymeric phosphates at -7 and -13 ppm respectively. MoP NMR signals were the only ones observed in the highly loaded molybdenum samples (53MoP). Furthermore, sample 53MoP exhibited a peak at -29 ppm which was due to AlPO4. This compound was not detected by XRD, suggesting that AlPO4 was present as crystals smaller than 3 nm or as an amorphous phase.
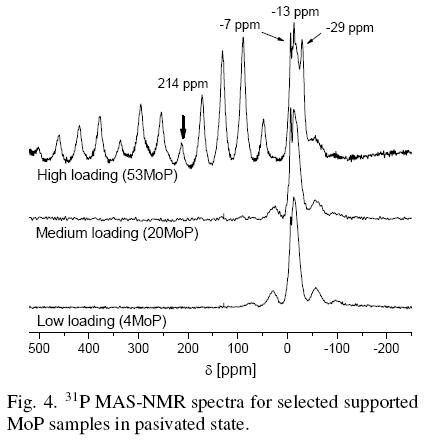
The phosphide NMR signals were not observed for low loading passivated samples. It seems that phosphides are easily oxidized to phosphate species. In fact, the spectra for samples with low and medium load exhibited peaks attributed to phosphate groups, revealing that these catalysts contained a mixture of mono and polyphosphates. Most probably, at high loading catalysts, the passivation step just oxidized a thin core of the MoP particles allowing MoP species to be detected by NMR. On the contrary, at low loading catalysts, the passivation step oxidized all the MoP particles due to its lower crystal size.
In order to disclose the evolution of phosphorous species, from the precursors to the catalysts, the 31P MAS-NMR spectra of the 13MoP (more active material) and 53MoP (high load) samples before reduction were recorded. Fig. 5 compares the spectra for the samples reduced at 1123 K without passivation. The two samples before reduction processes presented a single peak, characteristic of phosphate species (Fig. 5b and d). However, the peak for the sample loaded with the highest amount of metal and phosphorus (53MoP) was clearly shifted to a stronger field (-29 ppm) which must be attributed to the formation of AlPO4. The formation of AlPO4 and polyphosphates in P-modified alumina has been observed previously (Quartararo et al., 1999). The results indicated that the phosphide phase could be originated from different precursors depending on the Mo loading. Thus the 13MoP precursor has a mixture of mono and polyphosphates while the 53MoP precursor was probably a mixture of polyphosphates and AlPO4 species.
The samples reduced at 1123 K (Fig. 5, spectra a and c) showed NMR signals due to phosphide species; nonetheless some phosphates remained for both cases. Certainly, for sample 53MoP, the intense peak close to -29 ppm revealed the high thermal stability of aluminophosphate (AlPO4) in reduced conditions, which in its pure form may be stable up to 2073 K. Besides, NMR peaks of phosphates in the sample 13MoP, reduced at 1123 K, were less intense and less broad than the precursor (Fig. 5a). This result suggests that the phosphates are stabilized on the Al2O3 surface in this sample as shown as follows:
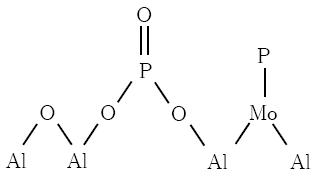
The 31 P NMR phosphide peaks were not observed for the 13MoP passivated sample (not shown here), but in the spectrum of the 13MoP reduced sample (Fig. 5a) they were clearly resolved. This result reveals that the passivation step transforms the entire MoP phase into phosphate species and it also suggests that particles in the 13MoP sample were smaller and more active for the oxidation reaction. Lastly, 31P NMR spectrum (not shown) for a reference sample containing 6 wt % of P before reduction at 1123 K shows a single peak at -29 ppm related to AIPO4 species.
Fig. 6 displays the 27Al MAS NMR spectra for the support and the selected MoP supported catalysts. The spectrum obtained for the carrier consisted of two peaks, at (20 and 7 ppm, due to tetrahedral and octahedral aluminum species, respectively. Spectra of the samples with low and medium Mo-P loading and samples with high Mo-P loading were similar, but in sample 53MoP an additional peak was observed at 40 ppm, attributed to the AlPO4 species (Vaneck et al., 1995). Sample 53MoP showed the signal corresponding to octahedral aluminum at 7 ppm, but an additional peak was resolved ca. -8 ppm. The two stronger field peaks in high Mo-P loading samples should be attributed to aluminum coordinated to six oxygen atoms to form octahedral species. Note that the support contains octahedral and tetrahedrally coordinated aluminum. The different strong field resonances observed should then be attributed to aluminum from the support reacted with phosphorous to form, for example, the amorphous AlPO4 (signal ca. -13 ppm). Then, the peak at a stronger field would be assigned to Al octahedrally coordinated in AlPO4. In the same way, 31P MAS-NMR spectra of samples 26MoP, 40MoP and 53MoP revealed the presence of an AlPO4 phase.
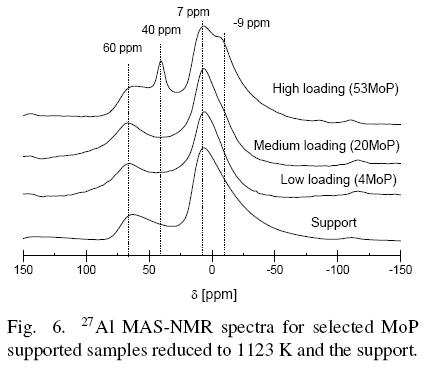
It can be concluded from the NMR results that AlPO4 was present when the P content was >6.5 wt%, as in the case of samples 26MoP, 40MoP, 53MoP, regardless of the Mo loading. The support used in this paper showed that the octahedral /tetrahedral aluminum ratio is similar to the ratio for y-Al2O3.
Several phosphorus oxo-compounds have been observed in hydrotreating catalysts depending on the phosphorus content. AlPO4 is only detected at phosphorus loadings higher than the theoretical monolayer coverage (Iwamoto and Grimblot, 1999). 31P and 27Al MAS NMR spectra showed that molybdenum phosphide was mainly detected in the 13MoP reduced sample and only a small amount of monophosphates and no AlPO4 phase were observed. For the 53MoP reduced sample, phosphide and several phosphates were detected, but AlPO4 dominated the phosphate signals (Fig. 5c). In this sample, the passivation step produced partial transformation from molybdenum phosphide to a core of phosphates. Table 3 shows the 31P MAS NMR deconvolution results for 13MoP, 53MoP and bulk MoP samples. It is worth mentioning that the NMR signals of the metal phosphide and phosphates arise from different interactions. The first is determined by metallic interactions (Mo atoms), while the second is determined by chemical interactions (valence electrons). Thus, integrations reported in Table 3 are a rough approximation (Lima et al., 2006). The relative quantity of phosphates in the bulk MoP sample indicates that the majority of phosphorus in the sample is molybdenum phosphide. The reduction and reduction-passivation steps for the bulk MoP sample did not change the phosphate/phosphide ratio, indicating that the bulk MoP sample was not sensitive to the oxygen atmosphere, possibly because the high concentration of Mo and P produced molybdenum phosphide in the reduction step, which is highly stable under the passivation conditions. For supported samples, the oxygen clearly reacted with the phosphides to produce phosphates; however, phosphates were also present in the reduced samples without the passivation step as explained beforehand. Further reduction time or further increase in temperature could eliminate the unreactive phosphates but would also increase the crystal size due to sintering effects.
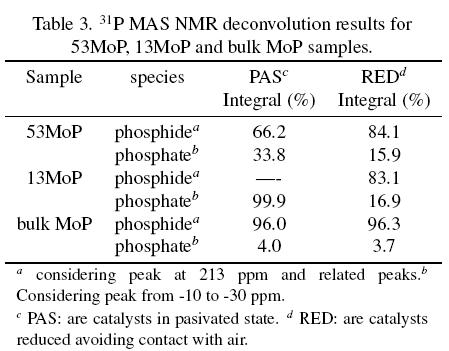
Fig. 7 shows the Temperature-Programmed Reduction (TPR) profiles for selected MoP/Al catalyst precursors with different Mo-P loadings. For the sake of conciseness we present the TPR analyses only for three representative samples, i.e. samples with low, medium and high loads levels of MoP were selected (4MoP, 20MoP and 53MoP respectively). For comparison purposes, the profile corresponding to the support (S) was also included.
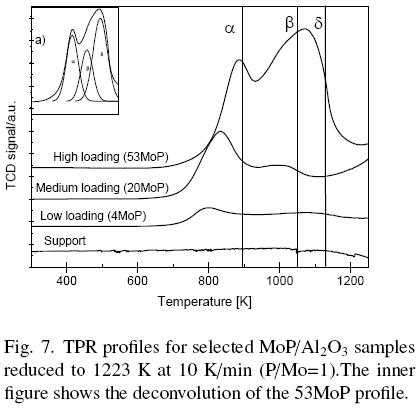
Three principal H2 consumption peaks were observed in the reduction processes between 750 and 1200 K, corresponding to the reduction of different species in the catalyst. No peaks were observed on the alumina support profile. All the samples, excluding the alumina support, exhibited a peak at low temperature designated by α and two peaks at high temperature designated by ß and δ (after deconvolution). Results indicated that the first peak (α) can be assigned to the reduction of MoO3 to MoO2, the second (ß) can be ascribed to the transformation from MoO2 to Mo and the third (δ) corresponded to the reaction of Mo with AlPO4 to form MoP (Mangnus et al., 1990). Moreover, XRD patters and NMR spectra were recorded after each step in the reduction process (not shown here) related to the TPR peaks and their reduction temperature. Mo, MoO3, MoO2, AlPO4 phases were detected in agreement with the proposed reduction steps, as in previous work by some of us (Montesinos-Castellanos et al., 2007). For low and medium loaded materials (4MoP and 20 MoP), the a peak was the main peak of the reduction profile. and peaks were prevalent in 53MoP but the ß peak was not clearly observed for low loaded catalysts, probably because the reduction of well dispersed species in this zone produced a pattern with only one peak at high temperature. The maximum for a, ß and δ peaks for the 53MoP catalyst were found at 883, 1000 and 1073 K, respectively. It is not surprising that the final step was the MoP formation from the reduction of AlPO4 and Mo0. It is well-known that the TPR of MoO3/Al2O3 produces Mo0 and that AlPO4 is reduced at high temperatures.
These results matched with those reported by Mangnus et al. (1990) and Montesinos-Castellanos et al. (2007). Three peaks were distinguished: the peak at low temperature (660-690 K) was attributed to the reduction of MoO3 and two peaks at high temperature were due to the reduction of Mo species and AlPO4. They concluded that the addition of P to the samples decreased the reducibility of the Mo species and that the TPR pattern of AlPO4 was quite similar to the Mo-P/Al2O3 sample with high P content. In our case, the P content corresponded to the P/Mo=1 atomic ratio, but the TPR profiles suggested that AlPO4 reduction was probably a crucial step in the synthesis of MoP catalysts.
It is known that Mo and P tend to produce phosphomolybdates and that this complex could be present in the catalysts, while Al-O-P and Al-O-Mo groups are also present. In our case, the dark blue color of the calcined precursor materials suggested that Mo and P produced molybdenum phosphate and Mo oxide species from phosphomolybdates in the calcination step. Besides, for samples such as 26MoP, 40MoP and 53MoP the presence of AlPO4 is expected due to the high P content. This explains the displacement of the peak toward higher temperatures at high loading sample. The 6 peak could also be explained considering that a complex formed by Mo and AlPO4 with poor reducibility requires a high temperature to finally produce the MoP phase. If AlPO4 were present in the low loading materials, the TPR pattern would show one peak at high temperature. The peaks in TPR profiles for low and medium loading samples could be explained by the simultaneous reduction of several species.
The reduction process of the samples with a high phosphorus content (such as 40MoP, 53MoP) showed a light yellow deposit on the bottom of the quartz reactor when the temperature reached that of the δ peak. This deposit could be due to P2, P4, or PH3 transported in vapor phase as reported by Mangnus et al. (1990).
The reduction step could be showing several species reducing at the same temperature (Oyama et al., 2009). The Mo and P content was evaluated by elemental analysis before and after reduction treatment and the results suggested that the composition range was MoP1-MoP0.82.
3.2 Morphological aspects
Three representative samples (bulk MoP, 13MoP and 53MoP) were studied by TEM and HRTEM. The micrograph for the bulk MoP sample (Fig. 8) shows agglomerated MoP particles with a mean size of 22 urn. Indexation of the diffraction patterns was performed and the measured interplanar distances were 0.319 nm (plane (001)), 0.280 nm (plane (100)), 0.213 nm (plane (101)), 0.161 nm (plane (110)) and 0.144 nm (plane (111)) of the MoP phase (JCPDS file 24-771). The diffraction pattern was consistent with the hexagonal MoP crystals. The morphology of bulk MoP consisted of small, regular-shape (spherical) particles with no other species being observed by electron diffraction, indicating that phosphates detected by 31P MAS-NMR are probably not crystalline.

For sample 53MoP, bright field and dark field techniques were used. Globular particles were also observed as for the bulk MoP sample. In this case, the particle size distribution was less uniform, where small crystals are mixed with large particles and the mean crystal size was larger than in the MoP bulk sample (Table 2).
The 53MoP sample after the passivation processes was also characterized by HRTEM (Fig. 9). Three different contrasts were observed. It was determined by fast Fourier transform, that the area with the strongest contrast was the crystalline MoP phase and the other two areas corresponded to amorphous phases. The passivation step was performed to prevent deep oxidation of the MoP producing a shell that could cover the particles. The passivated layer was observed for sample 53MoP; this layer, with a thickness of ~ 3.8 nm, covered all the MoP particles and most probably it was responsible for the phosphate species evidenced by 31P MAS-NMR. Sawhill et al. (2005) reported that Ni2P/SiO2 catalysts have a passivation layer with a thickness of ~2 nm; however, we found that in supported MoP, the thickness of the passivation layer can reach ~ 16 nm. In that case, the passivation process could transform most of the small particles of MoP into phosphate species, as in the 13MoP passivated sample.
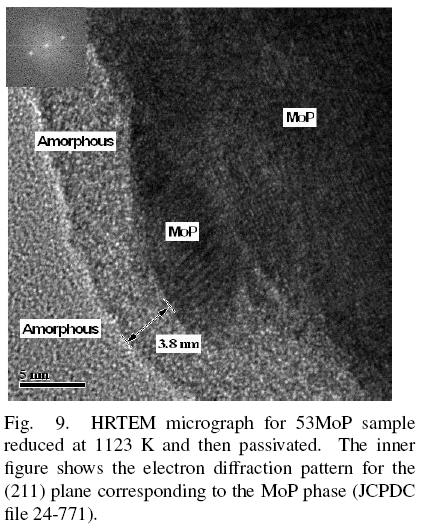
The TEM image of sample 53MoP (Fig. 9) shows several micrographs of MoP particles where a particular arrangement was observed: small particles (~ 6 x 15 nm) were deposited onto larger particles. MoP particles tend to form agglomerates in the reduction step, starting as small particles and growing as a function of the Mo and P concentrations in their nearness. Thus, all the particles would grow, some on the support and others would be formed on MoP particles already present on the surface.
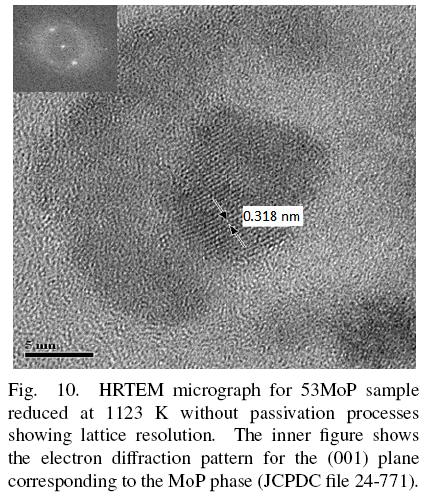
From HRTEM (Fig. 10), lattice parameters were measured for the 53MoP reduced sample the interplanar spacing was 0.318 nm corresponding to a (001) plane of molybdenum phosphide. This value was very close to those of MoP bulk sample. Therefore, the support did not affect the crystal structure of MoP significantly when high loadings were used, which is expected because it is hard to consider an interaction metal-support at very high loading of Mo. Fig. 10 also showed an amorphous area. This area was a compound formed by phosphorus and/or molybdenum. The TEM image with energy filtering revealed that Mo atoms and P atoms were evenly distributed in the particles; the amorphous vicinity in Fig. 10 was probably an unreduced amorphous AlPO4 phase near to MoP particles.
Using conventional TEM, it was not possible to determine clearly the particle size and morphology of MoP phase in sample 13MoP. The particles observed were very small and even when using the appropriate software a very large error was associated to measurements. Nevertheless, conventional TEM images (not shown here) illustrate that the crystal size in low loading sample 13MoP is smaller than in sample 53MoP. Moreover, the MoP particles form well-dispersed phases and that the tendency to form agglomerates or large particles decreased. This observation agrees with XRD and NMR results. The Energy-filtered Transmission Electron Microscopy (EFTEM) technique was used for sample 13MoP as well. The bright field image where several particles were observed is presented in Fig. 11a. In Fig. 11b, the Gatan Imaging Filter was adjusted for the Mo signal (Mo energy 35 eV) and the brighter areas correspond to the particles with strong contrast in Fig. 11a. Likewise, in Fig. 11c (filtering P signal 132 eV), the same intense areas can be observed. Thus, Mo atoms are completely associated with the P atoms. Moreover, MoP particles in sample 13MoP were small globular crystallites of ~4 nm in size that tended to form agglomerates. Intensity distributions from Fig. 11c showed that P atoms in MoP catalysts were close to Mo atoms and they did not remain dispersed on the entire support surface. Similar analyses were performed for sample 53MoP and led to the same conclusion.
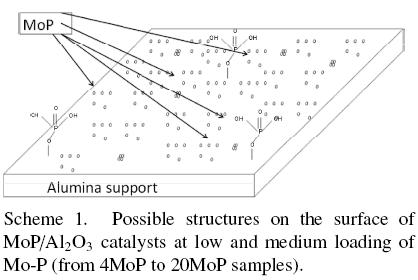
These results demonstrated that the morphology and the chemical species of MoP/Al2O3 depend on the molybdenum and phosphorus concentration. The amount of polymeric phosphates increased when phosphorus loadings increased. In agreement with these results, the scheme 1 and 2 model proposes an array of MoP and phosphate species on alumina for low and high loading of Mo and P. The principal species in the low loading catalysts were formed by monophosphates and small particles of MoP, as shown in scheme 1. In the high loading catalysts the principal species were polyphosphates, monophosphates, aluminium phosphate and large molybdenum phosphide particles. The quantity of the AlPO4 phase increases when phosphorus loading increases, being 16% of the total phosphates and phosphide present in the 53MoP catalyst, as mentioned earlier (Table 3).
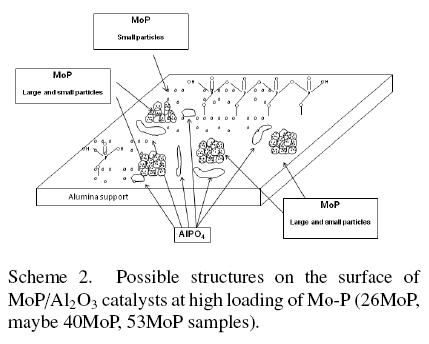
3.3 Textural aspects
The surface area of calcined support was 304 m2g-1 and the pore diameter of 7.8 nm, within suitable parameters for industrial HDS carriers. Table 1 reveals that specific surface area (SBET), total pore volume (TPV) and pore diameter (Ø) were parameters sensitive to molybdenum content. SBET decreased when the Mo and P content was increased. It should be highlighted that the values for textural properties for samples 3MoP to 20MoP were reasonable for HDS catalysts. SBET evolved from 304 m2/g (the support) to 27 m2/g for the sample containing the highest MoP loading. The SBET decreased dramatically when the content of Mo was as high as 20 wt%. These results could be explained by the fact that Mo-P species blocked the support pores, as one possibility. Very low TPV and SBET for sample 53MoP indicated that a high Mo and P loading leads to a catalyst with a very low surface area.
The highly loaded catalysts (26MoP, 40MoP, 53MoP) had a lower pore diameter than low metal loaded catalysts. The pore diameter for the support (S) was 7.8 nm and this value decreases in all the other catalysts, from 7.1 nm for 4MoP to 5 nm for 53MoP.
Iwamoto et al. (1999) reported that the effect of P on pore size distribution depends on catalyst preparation; for example, the impregnation method causes pore size to decrease with the P loading. Moreover, the high reduction temperature could result in sintering of the support, but in this case, the alumina was previously calcined at 1173 K. Decrease of pore volume or surface area could be due to the impregnation solution with low pH (the lowest value was 5.8).
3.4 Catalytic activity
MoP supported samples were tested in the hydrodesulfurization of DBT in a batch reactor and their activities were reported in terms of pseudo-first order rate constants for all catalysts. The results of these catalysts were compared with a conventional sulfided catalyst (10Mo-S). Fig. 12 shows that HDS activity increased with Mo loading reaching a maximum for the 13MoP catalyst. The rate constants of catalysts with a Mo content higher than 9.9 wt% decrease significantly. This behavior could be explained in terms of a lower dispersion for the 20MoP than that for the 13MoP sample, as revealed from the agglomeration of particles found by TEM and HRTEM. The average particle size of 13MoP and 20 MoP samples is 3 and 8.7 nm respectively. Moreover, the 26MoP (average particle size=22 nm), 40MoP (average particle size=39 nm) and 53MoP (average particle size=42 nm) samples showed that high loadings can be related to larger particle sizes and lower dispersions, as indicated for each catalyst. Besides, previously a relation between the fractal dimension of catalysts and their activities were reported, the higher fractal dimension was related with the lower catalytic activity. The fractal dimension increased with Mo loading (Montesinos-Castellanos et al., 2007).
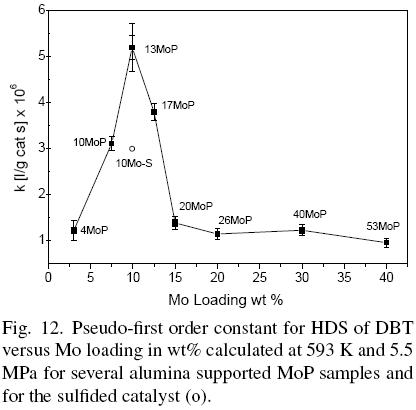
Results support that for low loading catalysts, the small and well-dispersed particles were responsible for the activity and that in high loading catalysts there is a mixture of small and large MoP particles, as concluded from HRTEM. The smaller MoP particles deposited onto the larger ones or on the support presumably were responsible for the activity.
The results suggest that only small particles were responsible for the catalytic activity. For low loading catalysts, the small particles were on the surface of the support and they corresponded to the most active catalysts. For high loading catalysts, high concentrations of Mo and P produced, in the reduction step, a very stable and sinterized phase with poor activity. Phillips et al. (2002) established that the activities of the catalysts decreased with the second pretreatment according to sulfidation < reduction < He degassing. According to the previous explanation, a second reduction step could produce growth for the MoP particles and this would explain to a certain extent why the He degassing pretreatment (just one reduction step) was better than a second reduction step.
On the other hand, the 13MoP catalyst was twice as active as the sulfided counterpart (10Mo-S) on the basis of catalyst mass. Nevertheless, several authors have reported larger differences between molybdenum phosphide and molybdenum sulfide with a different comparison basis or with a different model molecule. It has been reported that the M0P/SiO2 catalyst was four times more active than the sulfided catalyst with the same Mo loading in the HDS of thiophene. Nevertheless, molybdenum phosphide was compared with molybdenum sulfide after 120 h under reaction, where it was observed that the MoP catalyst increased their activity after 50 hours under reaction. Our result showed that the MoP/Al2O3 catalyst was more active than MoS2/ Al2O3 after only 8 hours under reaction conditions in the batch mode. The differences in activities compared with other authors could be due to differences in the systems and model compound used (Topsøe et al., 1996). Nevertheless, the fact remains that the molybdenum phosphide catalyst was more active than the sulfided catalyst with similar Mo loading. This result highlights the potential for these materials.
Regarding the identification of the active phases in metal phosphides, several papers focused on identifying such phases. Under reaction conditions some authors reported that phosphides transform into species formed by sulfur, molybdenum and phosphorus, i.e., a phosphosulfide. Furthermore, an increase in the reaction rate of the catalyst after a period of time, in a continuous reactor, has been interpreted as a change in the catalytic phase. However, this study does not allow us to obtain information about changes in the phosphide phase. In a batch reactor, sulfiding of the catalysts is less evident than in a continuous reactor, as has been the case in published works (Furimsky, 2003). Since no additional sulfur was added, the maximum concentration of sulfur in the batch reactor was 680 ppm as S in DBT, and at 15 % of conversion, it should have 1.62 x 10-4 mol of S for 1.87 x 10-4 of Mo (0.2 g of 13MoP). If a phosphosulfide has an S/Mo atomic ratio equal to 1, then the catalyst should have 86 % of Mo converted into phosphosulfide.
Conclusions
MoP supported on industrial alumina catalysts are suitable to catalyze HDS of DBT. Characterization techniques supported that MoP was not the only stable phase present on the support surface of the reduced materials; AlPO4, monophosphates and polyphosphates were also present. The AlPO4 phase was observed only in materials with P content > 6.5 wt%; also, polyphosphates were present mainly in highly loaded catalysts and monophosphates in low loaded catalysts. HRTEM and TEM showed that MoP particles formed agglomerates on the surface of the catalysts and those small particles were close to large particles for catalysts with high loadings. Energy filtering TEM demonstrated that Mo and P were very close to one another, which indicated that AlPO4, monophosphates and polyphosphates were close to MoP particles as shown by HRTEM results. A fraction of phosphorus impregnated produced MoP crystals on catalysts, while the rest of the phosphorous produced phosphates. A model is proposed for MoP-based catalysts with low and high Mo loadings.
The most active catalyst in HDS of DBT was the MoP sample containing 9.9 wt% of Mo. The reaction rate was not a function of Mo content. The small particles were more active than the large ones. The MoP catalyst was a more active catalyst when compared with MoS2 with the same Mo content. This result showed the potential of the materials described herein.
Acknowledgements
We would like to acknowledge the valuable help of Miss P. Castillo (electron microscopy, UAM-I) for the TEM micrographs. Thanks are due to PAPIIT-UNAM for financial support, grant IN107110.
References
Aegerter, P. A., Quigley, W. W. C., Simpson, G. J., Ziegler, D. D., Logan, J. W., McCrea, K. R., Glazier, S. and Bussell, M. E. (1996). Thiophene hydrodesulfurization over alumina-supported molybdenum carbide and nitride catalysts: Adsorption sites, catalytic activities, and nature of the active surface. Journal of Catalysis 164, 109-121. [ Links ]
Babich, I. V. and Moulijn, J. A. (2003). Science and technology of novel processes for deep desulfurization of oil refinery streams: A review. Fuel 82, 607-631. [ Links ]
Bridge, B. and Patel N.D. (1986). The elastic constants and structure of the vitreous system Mo-P-O. Journal of Materials Science 21, 1187-1205. [ Links ]
Clark, P., Wang, X. and Oyama, S. T. (2002). Characterization of silica-supported molybdenum and tungsten phosphide hydroprocessing catalysts by P-31 nuclear magnetic resonance spectroscopy. Journal of Catalysis 207, 256-265. [ Links ]
Clark, P. A. and Oyama, S. T. (2003). Alumina-supported molybdenum phosphide hydroprocessing catalysts. Journal of Catalysis 218, 78-87. [ Links ]
Euzen, P., Raybaud, P., Krokidis, X., Toulhoat, H., Le Loarer, J.-L., Jolivet, J.-P. and Froidefond, C. (2008). Alumina, in Handbook of Porous Solids, Edited by F. Schuth, 1591-1677, Wiley-VCH Verlag GmbH. [ Links ]
Furimsky, E. (2003). Metal carbides and nitrides as potential catalysts for hydroprocessing. Applied Catalysis a-General 240, 1-28. [ Links ]
Iwamoto, R. and Grimblot, J. (1999). Influence of phosphorus on the properties of alumina-based hydrotreating catalysts. Advances in Catalysis 44, 417-503. [ Links ]
Laredo, G. C., De los Reyes, J. A., Cano, J. L. and Castillo, J. J. (2001). Inhibition effects of nitrogen compounds on the hydrodesulfurization of dibenzothiophene. Applied Catalysis A-General 207, 103-112. [ Links ]
Lima, E., Bosch, P., Lara, V., Villarreal, E., Pina, C., Torres, G., Martín, S. and Leoín, B. (2006). Metal corrosion in bones implanted with Zinalco - A SAXS and NMR Study. Journal of Biomedical Materials Research Part B-Applied Biomaterials 76B, 203-210. [ Links ]
Mangnus, P.J., Van Veen, J.A.R., Eijsbouts, S., De Beer, V.H.J. and Moulijn J. A. (1990) Structure of phosphorus containing CoO-MoO3/Al2O3 catalysts. Applied Catalysis part A-General 61, 99-122. [ Links ]
McCrea, K. R., Logan, J. W., Tarbuck, T. L., Heiser, J. L. and Bussell, M. E. (1997). Thiophene hydrodesulfurization over alumina-supported molybdenum carbide and nitride catalysts: Effect of Mo loading and phase. Journal of Catalysis 171, 255-267. [ Links ]
Montesinos-Castellanos, A., Lima, E., de los Reyes, J. A. and Lara, V. (2007). Fractal dimension of MoP-Al2O3 catalysts and their activity in hydrodesulfurization of dibenzothiophene. Journal of Physical Chemistry C 111, 13898-13904. [ Links ]
Montesinos-Castellanos, A., Zepeda, T. A., Pawelec, B., Fierro, J. L. G. and de los Reyes, J. A. (2007b). Preparation, characterization, and performance of alumina-supported nanostructured Mo-phosphide systems. Chemistry of Materials 19, 5627-5636. [ Links ]
Montesmos-Castellanos, A., Zepeda, T. A., Pawelec, B., Lima, E., Fierro, J. L. G., Olivas, A. and de los Reyes-Heredia J.A. (2008). Influence of reduction temperature and metal loading on the performance of molybdenum phosphide catalysts for dibenzothiophene hydrodesulfurization. Applied Catalysis A-General 334, 330-338. [ Links ]
Nagai, M., Kiyoshi, M., Tominaga, H. and Omi, S. (2000). Molybdenum nitride as a long lifetime catalyst for hydrodesulfurization. Chemistry Letters 6, 702-703. [ Links ]
Oyama, S. T. (2003). Novel catalysts for advanced hydroprocessing: transition metal phosphides. Journal of Catalysis 216, 343-352. [ Links ]
Oyama, S. T., Clark, P., da Silva, V., Lede, E. J. and Requejo, F. G. (2001). XAFS characterization of highly active alumina-supported molybdenum phosphide catalysts (MoP/Al2O3) for hydrotreating. Journal of Physical Chemistry B 105, 4961-4966. [ Links ]
Oyama, S. T., Gott, T., Zhao, H. Y. and Lee, Y. K. (2009). Transition metal phosphide hydroprocessing catalysts: A review. Catalysis Today 143, 94-107. [ Links ]
Phillips, D. C., Sawhill, S. J., Self, R. and Bussell, M. E. (2002). Synthesis, characterization, and hydrodesulfurization properties of silica-supported molybdenum phosphide catalysts. Journal of Catalysis 207, 266-273. [ Links ]
Quartararo, J., Guelton, M., Rigole, M., Amoureux, J. P., Fernandez, C. and Grimblot, J. (1999). Sol-gel synthesis of alumina modified by phosphorus: a solid state NMR characterization study. Journal ofMaterials Chemistry 9, 2637-2646. [ Links ]
Sajkowski, D. J. and Oyama, S. T. (1996). Catalytic hydrotreating by molybdenum carbide and nitride: Unsupported MO2N and MO2C/Al2O3. Applied Catalysis A-General 134, 339-349. [ Links ]
Sawhill, S. J., Layman, K. A., Van Wyk, D. R., Engelhard, M. H., Wang, C. and Bussell, M. E. (2005). Thiophene hydrodesulfurization over nickel phosphide catalysts: effect of the precursor composition and support. Journal of Catalysis 231, 300-313. [ Links ]
Song, C. S. (2003). An overview of new approaches to deep desulfurization for ultra-clean gasoline, diesel fuel and jet fuel. Catalysis Today 86, 211-263. [ Links ]
Stinner, C., Prins, R. and Weber, T. (2000). Formation, structure, and HDN activity of unsupported molybdenum phosphide. Journal of Catalysis 191, 438-444. [ Links ]
Stinner, C., Prins, R. and Weber, T. (2001). Binary and ternary transition-metal phosphides as HDN catalysts. Journal of Catalysis 202, 187-194. [ Links ]
Stinner, C., Tang, Z., Haouas, M., Weber, T. and Prins, R. (2002). Preparation and P-31 NMR characterization of nickel phosphides on silica. Journal of Catalysis 208, 456-466. [ Links ]
Topsøe, H. B., Clausen, S. and Massoth, F. E. (1996) Hydrotreating Catalysis, Catalysis Science and Technology, Volume 11, Springer Verlag, Berlin, Germany. [ Links ]
Vaneck, E. R. H. , Kentgens, A. P. M. , Kraus, H. and Prins, R. (1995) A solid-state double-resonance NMR investigation of phosphorus-impregnated gamma-Al2O3. Journal of Physical Chemistry B99, 16080-16086. [ Links ]
Wu, Z. L., Sun, F. X., Wu, W. C., Feng, Z. C., Liang, C. H., Wei, Z. B. and Li C. (2004). On the surface sites Of MOP/SiO2 catalyst under sulfiding conditions: IR spectroscopy and catalytic reactivity studies. Journal of Catalysis 222, 41-52. [ Links ]
Zuzaniuk, V. and Prins, R. (2003). Synthesis and characterization of silica-supported transition-metal phosphides as HDN catalysts. Journal of Catalysis 219, 85-96. [ Links ]

