











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
Las malformaciones congénitas de las vías respiratorias bajas son infrecuentes. La incidencia anual estimada es de 30 a 42 casos por 100,000 habitantes o del 0.6 al 2.2% de todos los pacientes ingresados en hospitales generales1. El uso del ultrasonido prenatal permite un abordaje precoz, identificando la malformación en el 10% de los casos al nacimiento, mientras que el 14% son diagnosticados en la adolescencia en relación con un hallazgo radiológico o con síntomas crónicos de la vía aérea inferior2. El secuestro pulmonar es un segmento anormal de tejido broncopulmonar irrigado por una arteria anómala de origen sistémico. La malformación congénita de la vía aérea pulmonar (MCVAP), anteriormente llamada malformación adenomatoidea quística, es una alteración hamartomatosa del desarrollo pulmonar causada por el reemplazo de la pequeña vía respiratoria y el parénquima pulmonar por quistes. La asociación de ambas malformaciones es infrecuente. En la literatura internacional se han reportado aproximadamente 60 casos3.
Caso clínico
Se presenta el caso de una paciente de 4 meses de edad, hija de padres jóvenes, sanos, no consanguíneos. Antecedentes perinatales: madre de 30 años, producto de la primera gesta, embarazo normoevolutivo con adecuado control prenatal. Se realizaron seis ultrasonidos, y en uno de ellos se detectó una lesión sugestiva de secuestro pulmonar a la semana 24 de edad gestacional. La paciente fue sometida a tratamiento quirúrgico intrauterino, y en la semana 35 se reportó la resolución del secuestro. Se obtuvo producto único vía cesárea, sin complicaciones al nacimiento. Esquema de vacunación completo y adecuado desarrollo psicomotor.
La paciente fue evaluada por el servicio de neumología tras el antecedente de secuestro pulmonar, valorando la radiografía de tórax en proyección anteroposterior y lateral, colimada, que mostró tráquea levemente desplazada a la derecha, bien penetrada, con cinco espacios intercostales derechos e izquierdos, ángulos conservados, estructuras óseas y magras íntegras, silueta cardiaca desplazada a la derecha en proyección lateral con radioopacidad retrocardiaca hacia el segmento 8 (Figura 1). Ante este hallazgo, se ingresó como una malformación pulmonar en estudio, probable secuestro, para confirmación diagnóstica y manejo oportuno.
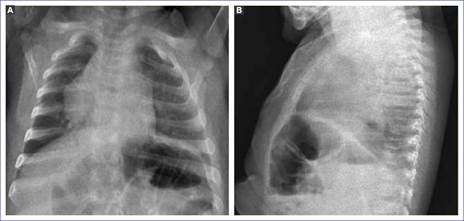
Figura 1 Radiografía de tórax. A: proyección posteroanterior en la que se evidencia la silueta cardiaca ligeramente desplazada a la derecha. B: radioopacidad hacia el segmento 8 del lóbulo inferior izquierdo en proyección lateral.
En la exploración física se encontró a la paciente alerta, activa, hidratada; normocéfala, con cabello bien implantado, sin masas; cuello cilíndrico, móvil, sin adenopatías; tórax simétrico, normoexpansible, sin tiraje; pulmonares con murmullo vesicular audible, sin estertores; corazón con ruidos cardiacos regulares, sin soplos; abdomen semigloboso, depresible, sin masas, megalias ni dolor; genitales externos adecuados para edad y sexo; extremidades simétricas, móviles, pulsos periféricos presentes, sin edema, sin acropaquias ni acrocianosis; piel con adecuada coloración y textura; neurológico sin datos de focalización ni de irritación peritoneal, Glasgow modificado 15/15.
En los estudios de laboratorio se obtuvieron los siguientes resultados: tiempo de protrombina 11.5 s, tiempo de tromboplastina parcial 30.4 s; hemoglobina 11.8 g/dl, hematocrito 36%, leucocitos 7,500/mm3, neutrófilos 22%, linfocitos 65%, monocitos 11%, eosinófilos 2%, eritrocitos 4.9 × 106/µl, plaquetas 336 × 109/l.
Se realizó una angiotomografía de tórax, en la que se encontró una imagen heterogénea en la ventana para parénquima pulmonar, corte axial y coronal, por la presencia de una forma ovoidea de aspecto quístico, con pobre realce al medio de contraste. El resto de parénquima mostró aumento de la densidad, excepto en la parte basal posterior izquierda. En fase arterial se observó un vaso nutricio proveniente de la aorta, segmento torácico, a nivel de la decimoprimera vértebra torácica, que rodea la lesión, y otro vaso que proviene de las venas pulmonares izquierdas (Figura 2).
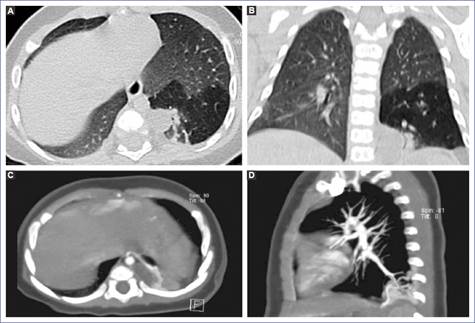
Figura 2 Angiotomografía. Ventana para parénquima pulmonar, corte axial (A) y coronal (B). Se muestra heterogéneo por la presencia de una imagen de forma ovoidea con dimensiones de 21 × 10 × 23 mm, aspecto quístico (5 unidades Hounsfield), que posterior a la administración de medio de contraste presenta pobre realce (19 unidades Hounsfield); se visualiza la opacificación. Resto del parénquima con aumento de la densidad, excepto en la parte basal posteroizquierda, donde se observan más hipodensos en relación con el parénquima restante. Fase arterial, corte axial (C) y sagital (D). Se observan un vaso nutricio proveniente de la aorta -segmento torácico a nivel de la decimoprimera vertebra torácica- que rodea la lesión y otro vaso que proviene de las venas pulmonares izquierdas.
Se realizó gammagrama pulmonar con macroagregados de albúmina marcada con tecnecio 99 para valorar la ventilación/perfusión de área pulmonar comprometida, en el que se observó un incremento en la perfusión de la región hiliar y basal derecha (Fig. 3). El servicio de medicina nuclear sugirió la realización de un gammagrama de eritrocitos marcados, el cual reportó datos sugestivos de secuestro pulmonar en la topografía del lóbulo inferior del pulmón izquierdo (Fig. 4). Posteriormente, se realizó toracotomía posterolateral y lobectomía con diagnóstico posoperatorio de secuestro pulmonar extralobar basal izquierdo. Se reportaron como hallazgos quirúrgicos vena subclavia izquierda permeable, secuestro extralobar supradiafragmático basal izquierdo con rama nutricia de aorta abdominal y quiste de pared torácica.
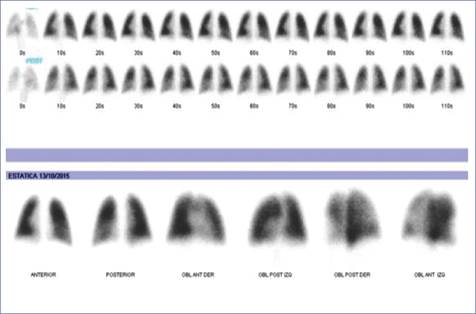
Figura 3 Gammagrama pulmonar con macroagregados de albúmina marcados con tecnecio 99. Incremento en la perfusión en la región hiliar y basal derecha (aumenta la captación de radiofármaco en fase dinámica).

Figura 4 Gammagrama de eritrocitos marcados. Datos sugestivos de secuestro pulmonar en topografía de lóbulo inferior del pulmón izquierdo. Silueta cardiaca lateralizada a la derecha de la línea media.
El estudio patológico del espécimen de lobectomía izquierda y de resección de quiste pulmonar reportó, en el lóbulo inferior del pulmón izquierdo, secuestro pulmonar extralobar con MCVAP tipo 2, enfermedad vascular pulmonar grado B de Rabinovitch y quiste broncogénico (Figura 5 y 6).
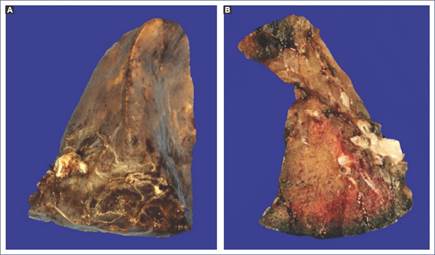
Figura 5 Descripción macroscópica. A: espécimen quirúrgico de forma triangular, de 41 g y 4 × 3.5 cm. La superficie externa está cubierta completamente por pleura, que es lisa, brillante, de color gris, en la porción basal del lóbulo. Quiste pulmonar: espécimen nodular de 1.5 × 1 × 0.3 cm, blando, de color blanco amarillento. B: al corte se observa un vaso sanguíneo de 0.3 cm de diámetro que penetra en el parénquima pulmonar; a nivel del hilio pulmonar se identifican un bronquio principal y vasos venosos con pared adelgazada. Al corte del quiste se observa una cavidad quística, cubierta por una pared de 0.3 cm de espesor.
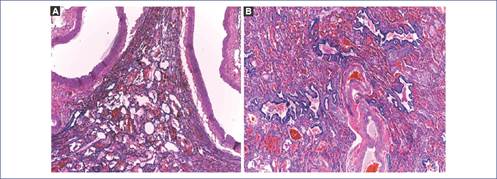
Figura 6 Descripción microscópica. A: los cortes muestran un hilio lobular con arteria, vena y bronquio, y la vena pulmonar con engrosamiento irregular por hiperplasia muscular. Arteria y bronquio sin alteraciones. B: el corte del vaso aberrante corresponde a una arteria con fibrosis irregular de la íntima e hiperplasia de la muscular. El parénquima adyacente se muestra con arquitectura anormal por la presencia de formaciones quísticas revestidas por epitelio cilíndrico ciliado, colapso y hemorragia intraalveolar. En el resto del parénquima se observan hemorragia reciente intraalveolar, sobredistensión de bronquiolos y alvéolos, y datos de enfermedad vascular caracterizados por hipertrofia de la media y aumento del tejido conectivo de la adventicia de arteriolas preacinares. El quiste presenta una pared muscular gruesa revestida por epitelio respiratorio. Se observan glándulas submucosas e islas de cartílago hialino en la pared.
La paciente tuvo una adecuada recuperación y evolución posterior al manejo quirúrgico. La radiografía de tórax después de un mes del procedimiento no mostró alteraciones aparentes (Figura 7).

Discusión
Existe un grupo de anomalías, que se han denominado malformaciones broncopulmonares del intestino anterior, que comprenden los quistes broncógenos, el secuestro pulmonar, los quistes de duplicación y las MCVAP3.
El secuestro pulmonar es una malformación de un territorio del pulmón que queda desconectado del árbol traqueobronquial y que es irrigado por una arteria sistémica, generalmente procedente de la aorta en lugar de la arteria pulmonar correspondiente. El origen corresponde probablemente a un trastorno de la vasculogénesis del área afectada o a una oclusión de los vasos que la deberían irrigar4),(5. Por otro lado, la MCVAP es un desarrollo hamartomatoso anormal con proliferación de bronquiolos que forman quistes en lugar de alvéolos. Se caracteriza por ser una masa multiquística con tejido pulmonar y proliferación de estructuras bronquiales, y se atribuye a un sobrecrecimiento de los bronquiolos con supresión del desarrollo alveolar6.
El secuestro pulmonar representa el 6% de las malformaciones pulmonares congénitasy se le estima una incidencia del 0.15-1.7%, mientras que la MCVAP constituye el 25% de todas las lesiones congénitas del pulmón3),(4),(7.
En lo referente a la clasificación, el secuestro pulmonar se divide en dos tipos: extralobar e intralobar. El secuestro intralobar se encuentra dentro del pulmón, cubierto por la pleura visceral, íntimamente conectado al pulmón adyacente. Se localiza en el lóbulo inferior en el 98% de los casos, y se asocia con hernia diafragmática, anomalías cardíacas congénitas y MCVAP6. Esta malformación se presenta en el 75% de los casos.
El secuestro extralobar es una masa de tejido pulmonar rodeada por su propia pleura que se presenta en el 25% de los casos8. En general, se localiza en los segmentos posteriores de los lóbulos inferiores. En el 90% de los casos se presenta en el lado izquierdo, además de que se localiza con mayor frecuencia en el tórax. En los casos restantes, la localización es intraabdominal, concretamente infradiafragmática y en la región suprarrenal izquierda9.
En 1977, Stocker y Drake10 clasificaron la MCVAP, según sus características clínicas y patológicas, en tres tipos: 1, 2 y 3. Posteriormente se adicionaron los tipos 0 y 4, según el lugar de origen de las lesiones11. Además, se introdujo el término MCVAP debido a las características distintas de cada tipo y a que las lesiones quísticas y adenomatoideas no estaban presentes en todos los tipos12:
-El tipo 0 consta de estructuras similares a bronquios que representan displasia acinar o agenesia; son raras y se asocian con malformaciones cardiovasculares. Resultan incompatibles con la vida.
-El tipo 1 comprende estructuras similares a bronquios y bronquiolos con grandes quistes. Pueden visualizarse en los primeros meses de vida, en niños y jóvenes adultos, y representan el 65% de los casos.
-El tipo 2 (10-15% de los casos) representa estructuras bronquiolares y acinares. Se observan durante el primer año de vida. Algunas tienen un pronóstico pobre debido a que están asociadas con anomalías incompatibles con la vida, como la agenesia renal. Los quistes miden aproximadamente 0.5-2 cm.
-El tipo 3 (5% de los casos) representa bronquiolos, conductos y sacos alveolares semejando estructuras medioacinares, que se visualizan como pequeños quistes de 0.2 cm o como estructuras sólidas. Este tipo se observa durante los primeros días o meses de vida. Se asocia con polihidramnios y anasarca fetal, y presenta una alta mortalidad.
-El tipo 4 (10-15% de los casos) es una malformación del acino distal que se observa como quistes de distribución periférica con apariencia hamartomatosa. Se diagnostica en recién nacidos y hasta los 4 años, y se ha observado que compromete un solo lóbulo en el 80% de los casos. Presenta una excelente sobrevida5.
Los signos y síntomas del secuestro pulmonar, tanto extralobar como intralobar, pueden pasar desapercibidos hasta el periodo neonatal, escolar o la adolescencia con la aparición de neumonías recurrentes13. Las lesiones expansivas de las MCVAP dan lugar a síntomas progresivos; si son de gran tamaño, pueden comprometer el desarrollo pulmonar en el periodo fetal, apareciendo pulmones hipoplásicos. Por efecto de masa, pueden impedir la adecuada contractilidad cardiaca y la compresión de la vena cava, con aparición de hidrops fetal. Si comprimen el esófago, al impedir la deglución del líquido amniótico aparecerá polihidramnios. Se han descrito también neumotórax de repetición, bronquitis obstructiva y dolor torácico, aunque son menos frecuentes14.
El diagnóstico se puede realizar desde la vida prenatal mediante ecografía, usualmente entre las semanas 18 y 24 de gestación. El secuestro pulmonar se visualiza en las ecografías prenatales como una masa hiperecogénica en el sector basal y posterior del hemitórax afectado. La ecografía Doppler color es útil para demostrar el vaso anómalo. En las radiografías de tórax, los secuestros extralobares se manifiestan como masas sólidas bien definidas adyacentes al mediastino o en el ángulo cardiofrénico, a veces subdiafragmáticas, aunque pueden confundirse con tumores mediastínicos. Los secuestros intralobares pueden contener aire, suelen ser más heterogéneos y menos definidos, y pueden imitar una neumonía o un absceso pulmonar13),(15. También pueden observarse bronquiectasias, atelectasia subsegmentaria, desplazamiento del mediastino y aumento de tamaño del hilio pulmonar homolateral. En las tomografías se puede observar una masa quística con contenido aéreo o líquido, enfisema focal e hipervascularización del tejido afectado. La presencia del vaso anómalo se demuestra con tomografía con contraste intravenoso, lo que evidencia una estructura lineal que nace de la aorta6.
Mediante ecografía, la MCVAP puede identificarse como una masa sólida o quística en uno de los pulmones fetales, o como un quiste dominante rodeado de quistes de menor tamaño. En la radiografía simple y en la tomografía se manifiestan como lesiones unilaterales con múltiples quistes aéreos redondeados y de paredes finas, variando el número y el tamaño de los quistes según el tipo14.
A la fecha, el tratamiento de elección del secuestro pulmonar y la MCVAP es quirúrgico. Sancho-Hernández, et al.16 reportaron que la capacidad de involución del secuestro pulmonar es pobre. Refieren que, al no existir indicadores pronósticos claros en la literatura actual sobre el potencial de malignidad e infección del secuestro pulmonar y la MCVAP, la conducta expectante permanece como una opción experimental. Por lo tanto, la cirugía electiva podría recomendarse hasta la lactancia temprana menor de 6 meses por el momento inmunitario favorable de este grupo de edad, más que por circunstancias estadísticas16. Otros autores, como San Vicente, et al.17, han referido que el manejo quirúrgico debe ser el de elección, aun en los casos asintomáticos, por el riesgo de recidivas; en su experiencia, presentaron un caso con degeneración maligna.
La asociación de secuestro pulmonar y MCVAP se ha descrito en aproximadamente 40-60 casos desde su primera observación en 19493),(18. La forma más frecuente de asociación es un secuestro intralobar junto con una MCVAP tipo 2. Por otro lado, Zangwill y Stocker19 reportaron que, de 30 casos de secuestro pulmonar extralobar, aproximadamente el 25% presentaban una MCVAP9. Conran y Stocker20 refirieron una coexistencia de secuestro pulmonar extralobar y MCVAP tipo 2 superior al 50% de los casos. La presencia conjunta de las dos malformaciones indica un defecto común en el desarrollo embriológico3),(19),(20. Ambas son consideradas lesiones híbridas, diagnosticadas solo por histopatología. Patológicamente, estas lesiones muestran datos histológicos combinados16.
En el presente caso, la paciente fue diagnosticada con secuestro pulmonar en la semana 24 de gestación. Recibió manejo durante esta etapa, aunque no se especificó en la historia clínica qué tipo de intervención se utilizó. La mayoría de los casos reportados tienen éxito en la resolución de la enfermedad, lo que evita procedimientos en la etapa posnatal. No hay un acuerdo sobre si la intervención debe realizarse intraútero o a los pocos meses de vida, aunque la mayoría de los autores concuerda en que, si existen lesiones extensivas o que producen hidrotórax poniendo en riesgo al feto, debe realizarse tratamiento prenatal21. Por otro lado, se han informado casos en los que se detienen el hidrotórax y la hidropesía, pero no cesa el crecimiento de tejido pulmonar anormal; en estos casos deben realizarse una toracotomía y la extirpación quirúrgica en el periodo neonatal temprano22.
La paciente referida en este caso no presentó ningún síntoma durante los primeros 4 meses de vida. El antecedente importante de cirugía fetal motivó a los padres a realizar el seguimiento por neumología, donde, además de secuestro pulmonar, en el estudio histopatológico se encontró una MCVAP en el interior del secuestro y un quiste broncogénico.
Por lo expuesto anteriormente, se considera que los pacientes con antecedentes prenatales de malformaciones pulmonares deben tener seguimiento neumológico desde la etapa neonatal, aun siendo asintomáticos. El diagnóstico prenatal de estas lesiones no es un predictor confiable de las características histopatológicas de las mismas, por lo que es necesario hacer un diagnóstico diferencial transoperatorio e histopatológico confirmatorio, y la búsqueda de afecciones asociadas en todos los pacientes, sintomáticos y asintomáticos16.
Identificar el tipo de malformación de manera acertada proporciona datos estadísticos valiosos para futuras investigaciones; además, ayuda a ofrecer un manejo oportuno y un adecuado seguimiento al paciente, y a proporcionar una mejor orientación a las familias.

