










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.62 no.5 México sep./oct. 2005
Artículo original
Síndrome de Klippel–Feil: una enfermedad musculoesquelética, con malformaciones cardiovasculares asociadas
Klippel–feil syndrome, a skeletal muscle disease associated to cardiovascular anomalies
Dr. Jesús de Rubens–Figueroa, Dra. Gabriela Zepeda–Orozco, Dra. Alma González–Rosas
Departamento de Cardiología, Instituto Nacional de Pediatría, Secretaría de Salud, México, D. F., México.
Solicitud de sobretiros:
Dr. Jesús de Rubens Figueroa,
Camino a Sta. Teresa 1055, S–09, Col. Héroes de Padierna, Deleg. M. Contreras,
C. P. 10700, México, D. F., México.
Fecha de recepción: 02–06–2005.
Fecha de aprobación: 13–10–2005.
Resumen
Introducción. El síndrome de Klippel–Feil (SKF) es un padecimiento de herencia autosómica dominante con penetrancia reducida y expresión variable, en el cual la tríada clínica inicial es de cuello corto, disminución en la movilidad del cuello e implantación baja del cabello posterior. Se asocia a otras malformaciones congénitas, entre ellas a cardiopatía (4–14%). El diagnóstico de la cardiopatía se debe realizar por el conocimiento de la asociación con este síndrome y con el apoyo del ecocardiograma.
Material y métodos. Estudio retrospectivo, lineal, descriptivo y observacional de los niños con SKF atendidos durante los últimos 22 años en el Instituto Nacional de Pediatría, SS. Se analizaron las anomalías cardiovasculares, género, edad, tipo de herencia, datos clínicos y evolución. El diagnóstico clínico se realizó por el ortopedista y el cardiológico apoyado por ecocardiograma.
Resultados. De los 46 pacientes, 24 correspondieron al género femenino y 22 al masculino. Las edades oscilaron entre un mes y 14 años.Todos los casos fueron de presentación esporádica. Las características clínicas más frecuentes fueron musculoesqueléticas, renales y cardiacas. Fueron valorados por el servicio de cardiología 19 pacientes, de los cuales 7 presentaron malformaciones cardiovasculares (7/46). Las cardiopatías que se presentaron fueron: comunicación interauricular (CIA), estenosis pulmonar (EP), comunicación interventricular (CIV) y dextrocardia, una paciente presentó además hipertensión arterial pulmonar primaria.
Conclusiones. La proporción de cardiopatías en el SKF en nuestro hospital fue de 0.5. Las cardiopatías más frecuentes fueron la CIA, CIV y la EP. Hasta el momento ninguno de los pacientes ha fallecido.
Palabras clave. Síndrome de Klippel–Feil; fusión de vértebras cervicales; cardiopatía congénita.
Abstract
Introduction. Klippel–Feil syndrome is a hereditary autosomal dominant disease with reduced penetrance and variable expression. It is characterized by multiple malformations but the 3 more constant are short neck, diminished mobility of the same and low implantation of posterior hair, associated to other congenital malformations as cardiopathy (4–14%).The diagnosis of cardiopathy must be done, first by knowledge of the association with this syndrome and after by clinical suspicion plus echocardiogram.
Material and methods. In a study of 22 years in the National Institute of Pediatrics, we found 46 cases with Klippel–Feil syndrome of which 7 had cardiovascular anomalies.These were diagnosed with echocardiogram.
Results. Of the 46 cases, 19 were evaluated by cardiologists; of these 7 (15%) had cardio–pulmonary malformations. Other malformations included: interauricular septal defect (ASD), interventricular septal defect (VSD), pulmonary stenosis (EP) and dextrocardia and one patient presented primary pulmonary hypertension.As of this communication, all patients are living.
Conclusion. In this series, the proportion of cardiovascular anomalies was 15%. We conclude that all patients with Klippel–Feil syndrome should have a thorough cardiological evaluation including an echocardiogram.
Key words. Klippel–Feil syndrome; abnormalities; congenital fusion of cervical vertebrae; heart defects, congenital.
Introducción
En 1912 Klippel y Feil descubrieron un paciente con malformaciones que se caracterizaban por: cuello corto y ancho, paladar hendido, implantación baja de cabello posterior y restricción del movimiento de cuello (debido a la fusión de dos o más vértebras cervicales).1–4
La frecuencia del síndrome de Klippel–Feil (SKF) ha sido estimada en 1 de cada 42 000 individuos. Hasta 1968 se habían publicado 505 casos de SKF en la literatura médica mundial, de los cuales 21 pacientes (4.2%) presentaron cardiopatía; sin embargo, en algunos pacientes no se buscó en forma intencional la asociación. Los informes posteriores han descrito esta asociación en porcentajes variables que van de 4 a 14%, incluyendo adultos.5 Es más frecuente en el género femenino que en el masculino con una relación de 1.5/1.6,7
La etiopatogenia corresponde a una alteración genética. Es un padecimiento de herencia autosómica dominante con penetrancia reducida y expresividad variable; el análisis cromosómico revela un cariotipo normal.Algunos casos son dominantes ligados al cromosoma X. El estudio de familias sugiere una transmisión autosómica recesiva y un gran número de casos son esporádicos.8 Hay una herencia heterogénea genética con deleciones descritas: 5q 11.2 y 8q 22.2; aunado a una mutación genética específica y factores ambientales, donde se ha identificado un locus genético. Esto explica la heterogenicidad del síndrome.9
La afección se debe a una alteración en la migración del tejido mesodérmico en el momento de la formación de los discos cervicales y del desarrollo de otros órganos y sistemas, en el mismo tiempo embriogénico, entre la tercera y cuarta semanas de desarrollo embrionario.5,10–13
La hipótesis de la secuencia considera una alteración inicial del tubo neural primitivo, lo cual explica la frecuente asociación de síntomas neurológicos.7 También se ha descrito la teoría de obstrucción vascular, donde hay secuencia de interrupción a nivel de la arteria subclavia, que explica la patogénesis del SKF.13
En la fusión tipo I se presenta una soldadura total de las vértebras cervicales hasta las superiores dorsales. En la tipo II se localiza a una o dos vértebras, generalmente acompañadas de fusión occípito–atlantoidea y de hemivértebras; es la más común, pero tiene mínimas manifestaciones clínicas. El tipo III se asocia la fusión cervical a un trastorno similar a nivel dorsal o lumbar, y en la tipo IV hay fusión cervical, torácica superior, dorsal inferior o lumbar.7,14
A la tríada inicial se asocian anomalías congénitas músculo–esqueléticas y de otros órganos como cerebro, riñón y corazón.
Dentro de las alteraciones músculo–esqueléticas se encuentran trastornos degenerativos y protrusión de los discos vertebrales, artrodesis posterior, osteofitos, siringomelia, estrechez de la unión cráneo–vertebral, escoliosis o xifosis, tortícolis por contractura muscular, pterigium coli, deformidad de Sprengel (incapacidad de la escápula para descender), tráquea corta, sindactilia, dedos supernumerarios, hipoplasia unilateral del músculo pectoral mayor, espasticidad, dolor e hiperreflexia, debilidad, parestesias y cefalea. Puede existir subluxación, degeneración vertebral o disminución del espacio intervertebral, que ocurre en forma espontánea.
También se ha asociado a este síndrome un gran número de alteraciones sistémicas: urinarias (agenesia renal, riñón en herradura); genitales como criptorquidia, ausencia de vagina y ovarios; pulmonares como estenosis de una rama de la arteria pulmonar y quiste broncongénico; gastrointestinales como duplicación intestinal, megacolon congénito y quiste neuroentérico; neurológicas secundarias a la lesión por inestabilidad occípito–cervical o atlanto–axial, hemiparesia, tetraparesia, ptosis, parálisis del séptimo par, compresión medular que puede causar dolor, debilidad, espasticidad, hiperreflexia de las extremidades superiores e inferiores, dolor en cuello, cefalea occipital. Puede asociarse a malformaciones del sistema nervioso central como: encefalocele, meningocele, hidrocefalia, malformación de Arnold–Chiari, microcefalia y siringomelia y alteraciones de la fosa posterior. Existen alteraciones neurosensoriales: sordera (30%);13 oculares: coloboma, microftalmía y ptosis palpebral.5,12,14,15 De las cardiovasculares, la malformación más frecuente es la comunicación interventricular (CIV); sin embargo, se han publicado numerosas asociaciones con diversas cardiopatías.7 En los tipos I y IV del SKF es donde se ha observado la asociación de malformaciones cardiacas.16
De los 21 casos de SKF asociados a cardiopatía (hasta 1968): cinco cursaron con CIV; uno con CIV y dextrocardia; uno con CIV asociada a comunicación interatrial (CIA) y pulmonar bicúspide; otro con CIV y arco aórtico a la derecha; tres con dextrocardia; dos con estenosis pulmonar (EP); dos con tronco común (TC); uno con ventrículo único (VU); uno con estenosis aórtica (EA); otro con defecto de la tabicación atrioventricular (D–AV); uno con isomerismo izquierdo; uno con doble cámara de salida de ventrículo derecho (DCSVD) con EP; y otro con conexión anómala de venas pulmonares (CAVP) asociado a arco aórtico derecho.10 En otro artículo se describen siete casos,de los cuales tres tuvieron persistencia del conducto arterioso (PCA); dos CIV; uno con arco aórtico a la derecha; y uno más con situs inversus.1 En 1979 se publicó la asociación con prolapso de la válvula mitral.10,17,18 En los últimos tiempos se han descrito asociaciones de este síndrome con cardiopatías más complejas como: CAVP, situs inversus, agenesia pulmonar con síndrome de Long–Ganon–Levin, con bifurcación carotídea intratorácica e incluso coartación de la aorta.14,19–21
Se han observado asociaciones con el síndrome de Turner, con la enfermedad de Von Recklinghausen o con ambas.22
El diagnóstico de SKF se basa en las manifestaciones clínicas descritas, la radiografía simple antero–posterior y lateral en la flexión y extensión de la columna cervical. Otros estudios de gabinete son la tomografía axial computada y la resonancia magnética.23
Algunos pacientes evolucionan asintomáticos los primeros años de la vida.
El diagnóstico diferencial se debe realizar con las siguientes alteraciones óseas: condrodisplasias, síndrome de Albright, displasias espondiloepifisiarias, metafisiarias y torácicas, síndrome de Wilderbanck, MURCS y VACTERR.14 Con algunas cromosomopatías como síndromes de Down, Turner, trisomía 4p, deleción 9p; con las embriopatías hidantoínica, síndrome feto–alcohólico, mucopolisacaridosis y otros síndromes (CHARGE, Meckel, Gruber y Noonan).7
Las complicaciones del SKF son: alteraciones degenerativas, protrusión de los discos vertebrales, osteofitos, siringomelia y estrechez en el nivel de la unión cráneo–vertebral. Las deformidades cervicales descritas tienden a agravarse con el tiempo. Por otra parte, las torácicas crean, ante todo, trastornos psicológicos.17 Los pacientes con hipermovilidad del segmento cervical superior tienen riesgo elevado de presentar secuelas neurológicas, y la alteración al movimiento del segmento cervical bajo predispone a una enfermedad degenerativa. Se deben evitar actividades deportivas para no ocasionar lesiones en el cuello.1,24 Por lo general, aquellos pacientes con cardiopatía producen hipertensión arterial pulmonar (HAP).
El pronóstico es variable en cuanto a las funciones del niño y va a depender de los problemas neurológicos. El pronóstico de la cardiopatía depende del tipo de ésta y el tiempo de evolución que influye en la presentación de complicaciones.
La educación familiar es vital para supervisar, mantener la función y prevenir el déficit neurológico por fusión, accidentes o trauma. En cuanto a la cardiopatía debe estudiarse cada caso para decidir el manejo médico o quirúrgico.
Material y métodos
En un estudio retrospectivo lineal, descriptivo y observacional, se revisaron los expedientes de niños con diagnóstico de SKF, con la finalidad de analizar las anomalías cardiovasculares. Se analizó género, edad, tipo de herencia, características clínicas y cardiovasculares, estudios de gabinete realizados y evolución. Se excluyeron los casos que no completaron los criterios diagnósticos (fusión de vértebras cervicales, disminución de la movilidad del cuello, cuello corto o implantación baja del cabello posterior), y se eliminaron pacientes con diagnósticos diferenciales como enfermedad facio–aurículo–vertebral (EFAV), Jarcho Levine (displasia espondilocostal, polidactilia y anomalías rectales), síndromes de Noonan, Turner y displasias costovertebrales.
Los pacientes con cardiopatía fueron diagnosticados mediante estudio ecocardiográfico.
Resultados
En la revisión de los últimos 22 años en el Instituto, se encontraron 46 casos, 24 del género femenino y 22 del masculino, las edades oscilaron entre un mes y 14 años, con una media de siete años; y mediana de cuatro años ocho meses de edad al momento del diagnóstico. En todos los casos el tipo de presentación fue esporádico, ninguno presentó dominancia ligada al cromosoma X, patrón autosómico recesivo o deleciones.
Las características clínicas más frecuentes fueron músculo–esqueléticas, renales y cardiacas. De las músculo–esqueléticas se presentaron: fusión de vértebras cervicales en 82.9%, cuello corto en 70.2%, hemivértebras cérvico–dorsales en 59.5%, y escoliosis cérvico–dorsal en 57.4% (Fig. 1). Otras manifestaciones fueron disminución en el movimiento del cuello, talla baja, anomalía de Sprengel, asimetría cuello–tórax, asimetría facial y retardo psicomotor (Fig. 2).
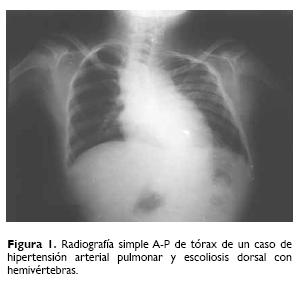
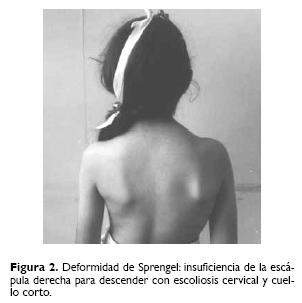
Las malformaciones renales se presentaron en 10 casos, siendo la más frecuente la hipoplasia renal en tres pacientes y las cardiacas en siete casos (Cuadro 1).
Hubo varias anomalías asociadas: síndrome hipotónico, luxación congénita de cadera, hipoplasia uterina, hipospadias, criptorquidia, hernia inguinal, quiste gastroentérico, quiste pilonidal, compresión del plexo braquial, costillas supernumerarias, pectus carinatum, parálisis de Erb, sindactilia, clinodactilia, neurofibromatosis y paladar hendido.
Los estudios de apoyo utilizados para el diagnóstico de SKF fueron radiografías de columna cérvico–dorso–lumbar, antero–posterior y lateral, otros con tomografía axial computada lineal y uno con resonancia magnética (Fig. 3).
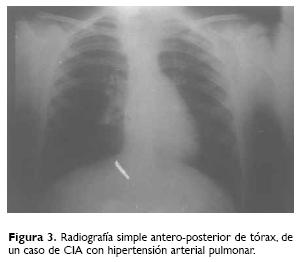
De los 47 pacientes, siete fueron dados de alta: dos postoperados de corrección de escoliosis y con mayoría de edad, dos asintomáticos sin requerir tratamiento, un postoperado de artrodesis cervical y de quiste entérico mediastinal con buena evolución, otro con artrodesis cervical y otro al cual se le ofreció cirugía de alto riesgo, sin ser aceptada por los familiares.
Dejaron de acudir en total 25 pacientes: 10 con estudios de extensión ortopédicos pendientes; 11 con estudios por realizar de otras especialidades; tres postoperados por ortopedia con buena evolución; y uno asintomático.
Acuden a control por la consulta externa hasta el momento 15 pacientes: cuatro con limitación de cuello y rehabilitación, cuatro en rehabilitación física y control por otros servicios, dos con ofrecimiento quirúrgico ortopédico, dos postoperados con osteotomía de barra cervical, dos con franca mejoría clínica y uno con manejo ortopédico conservador.
De los 46 pacientes, 19 fueron valorados por el servicio de cardiología, de los cuales 12 correspondieron a corazón sano y siete con malformaciones cardiovasculares: uno con EP, el cual presentó gradiente transpulmonar de 40 mm Hg, uno con CIA–ostium secundum de 10 mm de diámetro con cortocircuito de izquierda a derecha asociada a insuficiencia pulmonar leve y vena cava superior izquierda, dos con dextrocardia (sin malformaciones cardiacas), dos con CIV perimembranosas pequeñas con HAP de 65 y 84 mm Hg respectivamente, y uno con HAP primaria, que cursó con 45 mm Hg de presión sistólica pulmonar; todos diagnosticados por ecocardiografía, excepto este último al cual se le realizó el diagnóstico con angiocardiografía (Cuadro 2). No fueron valorados por el servicio de cardiología en total 27 pacientes, por no presentar sospecha clínica de malformación cardiaca.

De los siete pacientes con afección cardiovascular, en dos con dextrocardia asintomáticos no hubo necesidad de manejo cardiológico. Al paciente con HAP primaria con estabilidad hemodinámica se le da cita anual, con buena evolución clínica hasta el momento. Los dos casos de CIV cursaron con HAP, uno con 65 mm Hg de gradiente se operó de bandaje de la pulmonar en 1986 y se le efectuó corrección total en 1994, actualmente cursa con buena evolución, sin medicamentos. El otro paciente presentaba 84 mm Hg transpulmonar, se concluyó que la CIV funcionaba como válvula de escape a la presión pulmonar y se mantiene en vigilancia médica. La paciente con CIA continúa en vigilancia y se encuentra asintomática. En el paciente con EP, la válvula era trivalva, no displásica, y actualmente sin sintomatología.
Discusión
La tríada característica de inmovilidad de cuello, cuello corto e implantación baja del cabello posterior, secundaria a fusión de vértebras cervicales, se presentó en nuestros pacientes en un alto porcentaje. De la tríada, la implantación baja del cabello fue la de menor presentación (7 de 46 casos).
La incidencia que hay en la literatura de cardiopatías en el SKF es de 4 a 14% y se refiere que muchos casos no son valorados por el médico cardiólogo.1,18,23 En el estudio que se realizó, la proporción fue 0.15, la cual está dentro del límite superior de lo esperado. Se considera que podría ser mayor, siempre y cuando los médicos generales, pediatras y cardiólogos estuvieran más sensibilizados a valorar el área cardiológica de estos pacientes; esto se manifiesta claramente en nuestra casuística, al no haber valorado cardiológicamente a más de 50% de los 46 pacientes, por no haber manifestaciones clínicas de cardiopatía. De las siete alteraciones cardiovasculares que se presentan aquí asociadas a este síndrome, todas han sido publicadas con anterioridad.10,18,20
Por lo general, las cardiopatías que se asocian al SKF son benignas, esto quiere decir que pueden sobrellevarlas por varios años en aceptables condiciones físicas. Las cardiopatías congénitas en este estudio, correspondieron a la tercera malformación en frecuencia de este síndrome, sólo detrás de las músculo–esqueléticas y renales.
Como conclusiones, se pueden mencionar:
1. La proporción en el SKF de alteraciones cardiovasculares pulmonares es de 0.15, aun cuando muchos de los casos no fueron valorados por el servicio de cardiología (misma situación que sucede en otras publicaciones).
2. Todos estos pacientes deben ser atendidos por diversos especialistas, incluyendo cardiólogos, con o sin manifestaciones clínicas que hagan sospechar cardiopatía.
3. Las malformaciones cardiovasculares correspondieron a la tercera alteración en este síndrome, superadas por las músculo–esqueléticas y las renales.
4. Las cardiopatías más frecuentes fueron: CIV, CIA y EP.
5. Se recomienda que a todos los pacientes diagnosticados con SKF se les realice un ecocardiograma, estudio ideal para el diagnóstico de cardiopatías.
Referencias
1. Hesinger RN, Long JR, Mac Ewen GD. Klippel–Feil syndrome: a constellation of associated anomalies. J Bone Joint Surg. 1974; 56: 1246–53. [ Links ]
2. Berhman EW, Kliegman MR,Arvin MA. Nelson textbook of pediatrics. 15th ed. Philadelphia: WB Saunders; 1996. p. 1950. [ Links ]
3. Hikade KR, Bitar GJ, Edgerton MT, Morgan RF. Modified 2–plasty repair of webbed neck deformity seen in Turner and Klippel–Feil syndrome. Cleft Palate Craniofacial. 2002; 39:261–6. [ Links ]
4. Dattatraya MAG. Posterior cranial fossa dermoid in association with craniovertebral and cervical spinal anomaly: report of two cases. Pediatr Neurosurg. 2001; 35: 158–61. [ Links ]
5. Hinojosa M, Tatagiba M, Harada K, Samii M. Dermoid cyst in the posterior fossa accompanied by Klippel–Feil syndrome. Child Nerv Sust. 2001; 17: 97–100. [ Links ]
6. Mckusik VA. Mendelian inheritance in man, 10th ed. Baltimore: The John Hopkins University; 1992. p. 425–8. [ Links ]
7. Andersen GC. Atlas de síndromes pediátricos. 5a ed. México: Ed. Mc Graw Hill; 1989. p. 241–2. [ Links ]
8. Massuda H,Arikawa K,Yuda T.A total anomalous pulmonary venous connection associated with Klippel–Feil syndrome: a case report. Kyobu Geka. 1991; 44:417–20. [ Links ]
9. Tracy MR, Dormans JP, Kusumi K. Klippel–Feil syndrome: clinical features and current understanding of etiology. Cl Orthop Relate Res. 2004; 183–90. [ Links ]
10. Morrison GS, Perry WL, Scott PL. Congenital Brevicollis (Klippel Feil syndrome), and cardiovascular anomalies. Am J Dis Child. 1968; 115:614–20. [ Links ]
11. Algom M, Schesinger Z. Prolapse of the mitral valve in Klippel–Feil syndrome. Chest. 1981; 79: 127–8. [ Links ]
12. Allsopp GM, Griffiths S, Sgouros S. Cervical disc prolapse in childhood associated with Klippel–Feil syndrome. Childs Nerv Syst. 2001; 17: 69–70. [ Links ]
13. Franzen D, Sculte B, Beyer D, Neidel J, Koebke J, de Vivie R. Klippel–Feil syndrome associated with aortic coarctation. Cardiovascular Pathol. 2003; 12: 115–7. [ Links ]
14. Mc Gaughan JM, Kuna P, Das V.Audiological abnormalities in the Klippel–Feil syndrome. Arch Dis Child. 1998; 79: 352–5. [ Links ]
15. Oertel J, Piek J, Müller JU, Vogelgesang S, Warzok R, Gaab RM. Posterior fossa squamous cell carcinoma due to dedifferentiation of a dermoid cyst in Klippel–Feil syndrome. J Neurosurg. 2002; 97: 1244. [ Links ]
16. Clarke RA, Catalan G, Diwan AD. Heterogeneity in Klippel–Feil syndrome: a new classification. Pediatr Radiology. 1998; 28:967–74. [ Links ]
17. Udoshi MB, Shah A, Fisher VJ, Dolgin M. Incidence of mitral prolapse in subjects with thoracic skeletal abnormalities. A prospective study. Am Heart J. 1979; 7: 303–1 1. [ Links ]
18. Nora JJ, Cohen M, Maxwell GM. Klippel–Feil syndrome with congenital heart disease.Am J Dis Child. 1961; 102:858–64. [ Links ]
19. Al–Rajeh S, Chowdhary UM.Thoracic disc protrusion and situs inversus in Klippel–Feil syndrome. Spine. 1990; 15: 1379–81. [ Links ]
20. Baghat R, Pant K, Sing VK, Pant C, Gupta A, Jaggi OP. Pulmonary developmental anomaly associated with Klippel–Feil syndrome and anomalous atrioventricular conduction. Chest. 1992; 101: 1157–8. [ Links ]
21. Gailloud PH, Murphy KJ, Rigamonti D. Bilateral thoracic bifurcation of the common carotid artery associated with Klippel–Feil anomaly. Am J Neuroradiol. 2000; 21:941 –4. [ Links ]
22. Guille TJ, Miler A, Bwen RJ, Forlin E, Caro AP.The Natural history of Klippel–Feil syndrome clinic roentengenographic and magnetic resonance findings at adulthood. J Pediatr Orthop 1995; 15:617–26. [ Links ]
23. Mc Bride WZ. Klippel–Feil syndrome. Am Fam Physician. 1992; 45: 633–5. [ Links ]
24. Pizzutillo PD,Woods N, Nicholson L, Mac Ewen GD. Risk factors in Klippel–Feil syndrome. Spine. 1994; 19:2100–16. [ Links ]

