










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de cardiología de México
versión On-line ISSN 1665-1731versión impresa ISSN 1405-9940
Arch. Cardiol. Méx. vol.71 no.4 Ciudad de México oct./dic. 2001
Investigación básica
Importancia de la enzima convertidora de angiotensina (ECA) en la circulación coronaria
Importance of angiotensin converting enzyme in coronary circulation
Karin Mauer Díaz,* José Emilio Exaire Murad,** Bruno Alfonso Escalante Acosta*
* Departamento de Biomedicina Molecular del Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, México D.F.
** Hospital Médica Sur, México D.F.
Correspondencia
Bruno Alfonso Escalante Acosta.
Departamento de Biomedicina Molecular del CINVESTAV-IPN
Avenida Instituto Politécnico Nacional No. 2508, Colonia San Pedro Zacatenco, 7360 México D.F.
Tel: 57477000 Ext 5458.
Recepción: 22 de marzo de 2001
Aceptado: 25 de junio de 2001
Resumen
A nivel cardiovascular, se sugiere que la síntesis de angiotensina II por la enzima conversora de angiotensina es secundaria a otras vías como la de la quimasa. En este estudio valoramos la importancia de la enzima conversora de angiotensina en la circulación coronaria durante el desarrollo de hipertensión. Se estimularon corazones de ratas normales (n = 4), hipertensas (n = 4) e hipertensas tratadas con ramipril (n = 4), en un sistema de perfusión tipo Langendorff. La angiotensina II produjo vasoconstricción coronaria dependiente de la concentración, y el efecto fue mayor en corazones de ratas hipertensas comparado con ratas normotensas (p < 0.05). La angiotensina I también produjo vasoconstricción y el efecto fue mayor en corazones de ratas hipertensas en comparación a la respuesta de ratas normotensas (p < 0.05). El ramiprilat inhibió 78% de la vasoconstricción inducida por angiotensina I en corazones de ratas normotensas y 82% en ratas hipertensas (p < 0.05). El bloqueo de la enzima conversora de angiotensina in vivo, potenció el efecto vasoconstrictor de la angiotensina I en corazones de ratas normotensas e hipertensas (p < 0.05). El efecto inhibitorio del ramiprilat disminuyó significativamente (p < 0.05) cuando se bloquearon los receptores de bradicinina. En conclusión, la síntesis de angiotensina II en corazones de ratas normotensas e hipertensas depende prioritariamente de la enzima conversora de angiotensina. La inhibición de la enzima conversora de angiotensina in vivo pudiera inducir otras vías que generan angiotensina II y posiblemente la actividad de bradicinina determina parcialmente el efecto de la inhibición de la enzima conversora de angiotensina a nivel coronario.
Palabras clave: Angiotensina. Circulación coronaria. Enzima conversora de angiotensina.
Abstract
It has been suggested that angiotensin II can be synthesized by other enzymatic pathways besides angiotensin converting enzyme. We evaluated the importance of angiotensin converting enzyme in the coronary circulation during the development of hypertension. Hearts obtained from normotensive (n = 4) and hypertensive rats (n = 4) as well as from hypertensive rats treated with ramipril (n = 4) were stimulated with either angiotensin II or angiotensin I. In a Langendorff perfusion system, angiotensin II induced a greater dose-dependent coronary vaso-constriction in the hearts of hypertensive rats than in normotensive rats (p < 0.05). Furthermore, angiotensin I also induced coronary vasoconstriction, which was greater in the hearts of hypertensive rats than in normotensive rats (p < 0.05). Acute angiotensin converting enzyme inhibition reduced angiotensin I-induced vasoconstriction by 78% in the hearts of normotensive rats and by 82% in the hypertensive rats (p < 0.05), whereas in vivo angiotensin converting enzyme inhibition potentiated angiotensin I-induced vasoconstriction in the hearts of normotensive and hipertensive rats (p < 0.05). Bradykinin receptor's blockade decreased ramiprilat's inhibitory effect on angiotensin I-induced vasoconstriction (p < 0.05). Thus, the present study suggests that, in coronary circulation, angiotensin II synthesis is mainly angiotensin converting enzyme dependent. However, chronic in vivo inhibition could favor induction of other enzymes involved in angiotensin II synthesis. Evenmore, it is possible that the effect of angiotensin converting enzyme inhibition in coronary circulation depends on bradykinin activity.
Key words: Angiotensin. Coronary circulation. Angiotensin converting enzyme.
Introducción
El sistema renina-angiotensina (SRA) consiste en una cascada de interacciones entre enzimas y sustratos que culminan con la producción de angiotensina (Ang) II, el péptido activo responsable de los efectos conocidos de este eje bien difundido en todo el organismo.1
La síntesis de Ang II depende fundamentalmente de la conversión de Ang I en Ang II2 a través de la activación de la enzima conversora de Ang (ECA). Sin embargo, hay evidencias experimentales que sugieren la existencia de vías alternas, independientes de ECA, que contribuyen a la producción de Ang II en diversos tejidos a partir de angiotensinógeno y Ang I. Reportes recientes han demostrado la presencia de la enzima generadora de Ang II sensible a quimostatina o CAGE, el factor activador del plasminógeno tisular y la catepsina G entre otros.3-5 En el corazón se identificó una quimasa de alta eficiencia.6,7 Sin embargo, la especificidad de la actividad de esta enzima en el corazón es controvertida. En la circulación coronaria, la importancia de las diferentes vías enzimáticas implicadas en la conversión de Ang I a Ang II es variable dependiendo de la especie estudiada. Se ha descrito una enzima parecida a la quimasa con actividad dominante en todas las especies excepto en humano, conejo y cerdo.8 Hay estudios que sugieren que algunas quimasas de mamíferos tienen una especificidad amplia de sustrato y no generan Ang II.9 La existencia de estas vías alternas de síntesis de Ang II sugeriría que los fármacos inhibidores de la ECA (IECA), pudieran tener un efecto transitorio o poco potente.
En estudios clínicos y experimentales se observan acciones locales adicionales de los IECA, no relacionadas a la reducción en la presión arterial, tales como la regresión de la hipertrofia del músculo cardiaco, y que no pueden ser explicadas solamente por el bloqueo del SRA.10 Aún más, se ha descrito un papel importante de los inhibidores de ECA en la prevención de eventos isquémicos y aterogénesis.11
Hoy en día se sabe que la ECA es idéntica a la quininasa responsable del metabolismo y la degradación de la bradicinina (Bk).12 El bloqueo de la ECA por lo tanto aumenta en forma considerable la concentración local de Bk en todos los órganos.13 La potenciación del sistema de las quininas endógenas pudiera ser el responsable de los efectos adicionales de los IECA.14
Estos datos sugieren que en el tejido cardiaco, la ECA puede representar el principal mecanismo de generación de Ang II, por lo que los efectos terapéuticos de los IECA pudieran explicarse mediante la disminución en los niveles de Ang II y el aumento en la síntesis de Bk. En el presente trabajo nos propusimos como objetivo valorar la importancia de ECA en la circulación coronaria de ratas durante el desarrollo de hipertensión secundaria a coartación aórtica a nivel de la arteria renal.
Material y métodos
Cirugía de coartación aórtica. En este modelo experimental se emplearon ratas Wistar macho con un peso entre 200 y 400 gramos. Los animales se anestesiaron con éter inhalado. Previa antisepsia se realizó una incisión abdominal a nivel de línea media y se disecó por planos hasta entrar a cavidad peritoneal. Se rechazaron los intestinos con una gasa y se identificaron las estructuras vasculares. A nivel aórtico, entre ambas arterias renales, se colocó una ligadura (seda 3-0) creando una obstrucción parcial del vaso, para provocar una coartación aórtica artificial, dejando un flujo sanguíneo constante equivalente al diámetro de una aguja 21 a 22 G. Posteriormente se retiró el instrumental quirúrgico y se procedió a cerrar la pared abdominal por planos con sutura no absorbible.
Medición de la presión arterial sistólica (TAS). Después de 21 a 25 días de postoperatorio con alimentación y actividad normales se dejó a las ratas, ya hipertensas, en ayuno durante 24 hrs previas a la medición de TAS y a la disección del corazón. Cada animal se anestesió con pentobarbital sódico (35 mg/kg vía intraperitoneal) y se realizó una incisión en cuello a la altura de línea media, se disecó por planos y se localizó el paquete neurovascular. Se separó a la arteria carótida primitiva de las estructuras musculares y nerviosas adyacentes y se ligó distalmente con seda 3-0. En el lado proximal se colocó una pinza para interrumpir el flujo vascular y se realizó una pequeña incisión por la cual se canuló la arteria. Después de reiniciar el flujo arterial la cánula se conectó a un trasductor de presión, el cual nos dio una lectura de la TAS durante un periodo de 15 a 30 minutos.
Modelo de corazón aislado perfundido. Se hizo una incisión abdominal a nivel de línea media. Se disecó por planos hasta localizar la vena cava inferior a través de la cual se inyectó heparina (500 UI vía intravenosa) para lograr un efecto anticoagulante. Después de unos segundos se exsanguinó al animal por la misma vía, hasta extraer aproximadamente 8 a 10 mL de sangre. Se procedió a abrir tórax cortando el diafragma y ambas parrillas costales exponiendo así al corazón. Se disecó el pericardio y se realizó una pequeña incisión a nivel de aorta ascendente por arriba de la válvula aórtica, a través de la cual se canuló retrógradamente el corazón. Se cortaron las estructuras de la cara posterior para liberarlo y colocarlo en una caja de Petri con solución Ringer-Locke (NaCl 157.4 mM, KCl 5.63 mM, Glucosa 5.55 mM, NaHC03 1.78 mM, CaCl2 2.09 mM, pH 6.4, a 37°C, con burbujeo continuo de una mezcla de O2 y CO2 (95:5)) fría. Se fijó el corazón a la cánula y se aseguró que ésta quedara por arriba de la válvula aórtica cerrada para garantizar que el flujo fuera directamente al lecho coronario. Así se perfundió el corazón con la misma solución mediante una jeringa para insulina, hasta eliminar completamente los residuos de sangre a nivel de coronarias. Después se acopló el corazón al sistema de perfusión tipo Langendorff, y se perfundió mediante el bombeo continuo de solución Ringer-Locke. Al corazón se le adaptó un marcapaso externo estimulando con pulsos cuadrados continuos: 3 latidos/segundo, 2 voltios de amplitud, 200 mseg de duración y 200 mseg de retraso entre cada pulso. Bajo estas condiciones se permitió que el corazón se estabilizara durante 10 a 30 minutos. Se ajustó la presión media del sistema entre 70 y 85 mmHg como presión inicial en todos los grupos experimentales. Una vez estable la preparación se efectuaron las diferentes administraciones de Ang II o Ang I, obteniendo registros de cambios en la presión de perfusión coronaria.
Medición de cambios en la presión de perfusión coronaria. Para valorar el efecto vasoactivo de la Ang II y la Ang I sobre el corazón aislado perfundido se midió en el registro de presión de perfusión coronaria de cada experimento la diferencia entre la presión de perfusión media inicial y la presión de perfusión media máxima durante 40 minutos después de inyectar los diferentes fármacos al sistema, obteniendo así una delta de presión expresada en mmHg. La presencia de actividad por parte de la ECA en el lecho coronario se evaluó indirectamente midiendo respuestas de vasoconstricción coronaria inducidas por Ang I antes y después de preincubar la preparación con un inhibidor de la ECA (ramiprilat 1µM donado por AVENTIS PHARMA, México). Los experimentos también se realizaron en animales normotensos e hipertensos con tratamiento previo a base de ramipril 1 mg/kg/día por vía oral durante 7 días, el cual fue interrumpido de manera abrupta 24 horas antes de la preparación del órgano aislado. Para valorar el papel de Bk sobre el efecto de ramiprilat in vitro se preincubó la preparación con el antagonista selectivo de receptores de Bk HOE 140 (100 nM) (donado por AVENTIS PHARMA, México).
Grupos experimentales. Los grupos experimentales constaron de 4 animales cada uno (n = 4) y la medición de la TAS se basó en el resultado de 18 experimentos (n = l8). Como controles se usaron animales normales, no sometidos a cirugía de coartación aórtica y que representan a las ratas normotensas y en el grupo de animales hipertensos se incluyeron las ratas operadas, a las cuales se les realizó la coartación aórtica entre las dos arterias renales (Fig. 1).
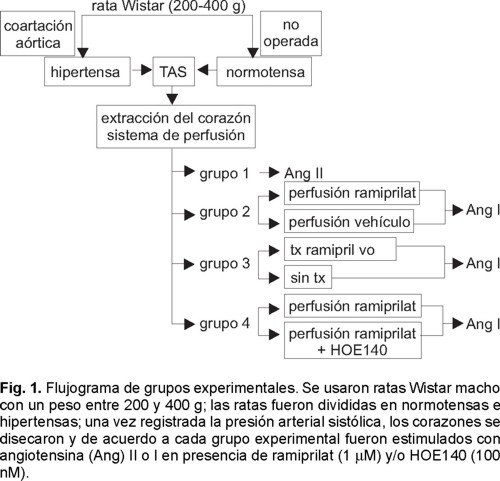
Grupo experimental 1: con el objeto de comparar el efecto vasoconstrictor de la Ang II en la circulación coronaria de ratas normotensas vs ratas hipertensas, se montaron corazones de ratas normotensas o hipertensas en un sistema de perfusión in vitro, y la circulación coronaria fue estimulada con concentraciones crecientes de Ang II (1, 2 y 8 ng). Los cambios en la presión de perfusión de la circulación coronaria se grafican como delta (Δ) en mmHg (Fig. 1).
Grupo experimental 2: con el objeto de demostrar la presencia de la ECA en la circulación coronaria, los corazones provenientes de ratas normotensas e hipertensas fueron montados en un sistema de perfusión in vitro y la circulación coronaria fue estimulada con Ang I (2 ng) en corazones perfundidos con vehículo o corazones perfundidos con el inhibidor de la ECA, ramiprilat (1 µM) durante 30 minutos antes de la administración de Ang I (Fig. 1).
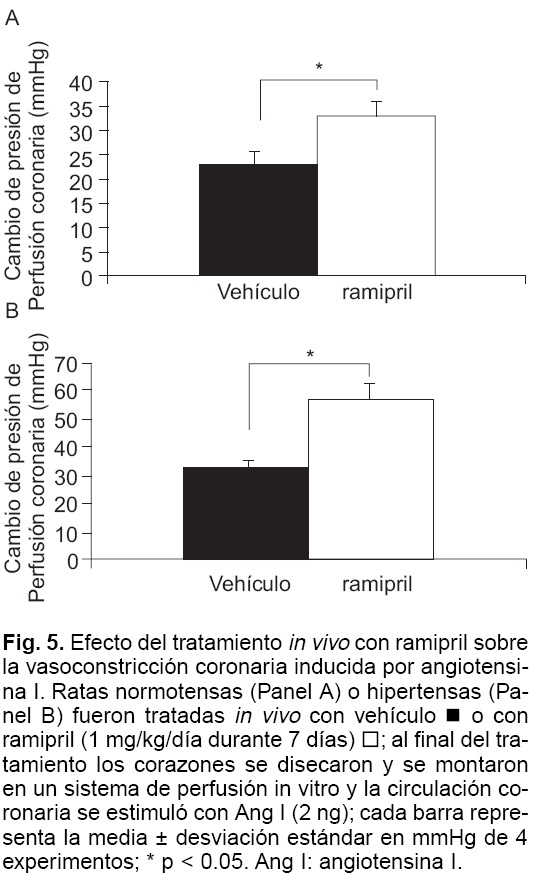
Grupo experimental 3: con el objeto de demostrar el efecto de la inhibición crónica de la ECA sobre la conversión de Ang I en la circulación coronaria, se trataron ratas normotensas o hipertensas durante 7 días con vehículo o ramipril (1mg/kg/día). Al final del tratamiento, los corazones se aislaron y se montaron en un sistema de perfusión in vitro y se estimularon con Ang I (2 ng) (Fig. 1).
Grupo experimental 4: con el objeto de demostrar la participación de la bradicinina (Bk) en el efecto del inhibidor de ECA en la circulación coronaria, se montaron corazones de ratas normotensas e hipertensas en un sistema de perfusión in vitro y después de perfundirlos durante 30 minutos con el inhibidor de ECA, ramiprilat (1 µM) ya sea en combinación con el antagonista de Bk HOE140 (100 nM) o en ausencia del mismo, se estimularon con Ang I (2 ng) (Fig. 1).
Análisis estadístico. Los datos obtenidos de cada serie experimental fueron sometidos a la prueba de análisis de varianza. Se consideró que había diferencias significativas cuando p < 0.05. Los resultados se presentan como la media ± la desviación estándar de 4 experimentos para cada grupo experimental.
Resultados
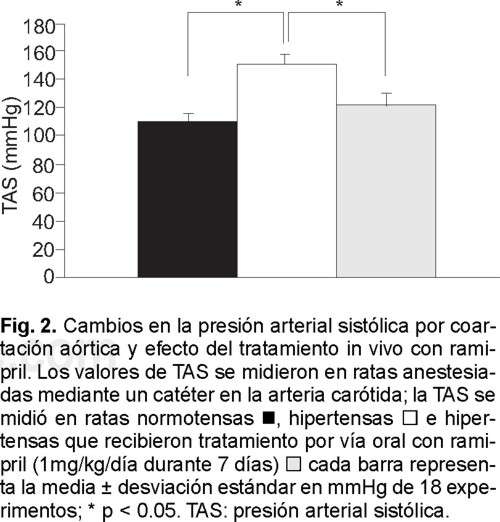
La presión arterial sistólica (TAS) de las ratas normotensas fue de 111 ± 5 mmHg, mientras que en las ratas hipertensas este valor se elevó a 153 ± 6 mmHg (p < 0.05). El tratamiento con ramipril (1mg/kg/día vía oral durante 7 días) disminuyó los valores de TAS de los animales hipertensos a 121 ± 6 mmHg, no habiendo diferencia significativa en comparación a las cifras de los animales normotensos (Fig. 2).
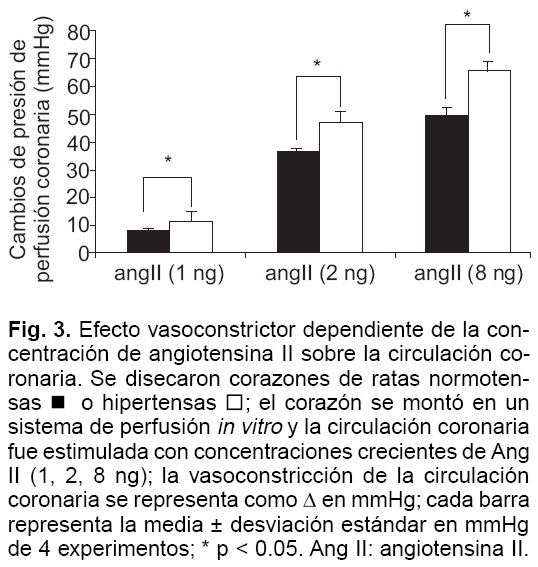
La aplicación de Ang II al sistema de órgano aislado generó vasoconstricción coronaria dependiente de concentración en los corazones de ratas normotensas e hipertensas. Ang II a las concentraciones de 1, 2 y 8 ng aumentó la presión de perfusión coronaria 7.75 ± 0.25, 36.5 ± 0.96 y 49.63 ± 2.98 mmHg respectivamente en corazones de ratas normotensas, mientras que en los corazones de ratas hipertensas las mismas dosis de Ang II aumentaron la presión de perfusión 11.88 ± 1.88, 47.13 ± 3.69 y 65.63 ± 3.29 mmHg respectivamente. Este efecto vasoconstrictor de Ang II fue mayor en los corazones de los animales hipertensos comparado con la respuesta en los animales normotensos (p < 0.05) (Fig. 3). Aún más, cuando el corazón se estimuló con 2 ng de Ang I, se observó un incremento en la presión de perfusión coronaria de 22.88 ± 1.81 mmHg en corazones de animales normotensos comparado con un aumento de 30.75 ± 1.49 mmHg en corazones de ratas hipertensas (p < 0.05).
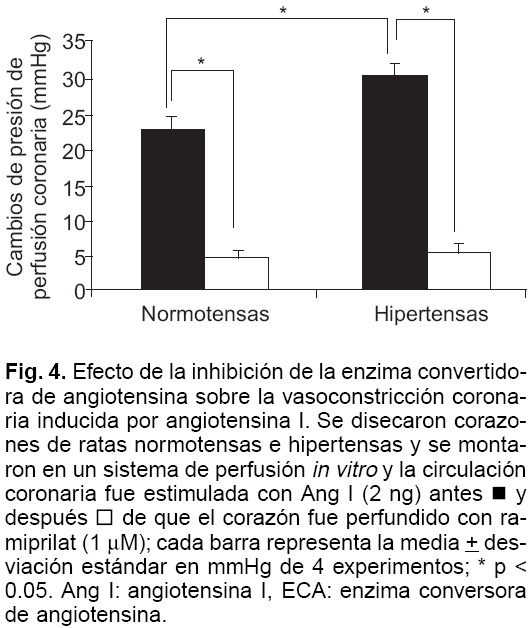
Con el objeto de evaluar la importancia de la ECA en la vasoconstricción inducida por Ang I, se probó el efecto de inhibir la ECA con ramiprilat. La presencia de ramiprilat (1 µM) en la solución de perfusión del corazón aislado inhibió significativamente la vasoconstricción coronaria inducida por Ang I en ratas normotensas e hipertensas, de tal forma que el efecto vasoconstrictor de Ang I se inhibió 78% en ratas normotensas y 82% en ratas hipertensas (p < 0.05) (Fig. 4).
El tratamiento in vivo con ramipril (1 mg/kg/día vía oral durante 7 días), el cual se suspendió 24 horas antes de montar la preparación, claramente potenció el efecto vasoconstrictor de Ang I tanto en corazones de ratas normotensas como en los de ratas hipertensas. El efecto vasoconstrictor de Ang I (2 ng) fue de 32.38 ± 2.48 mmHg en ratas normotensas y de 55.25 ± 5.92 mmHg en ratas hipertensas (p < 0.05) (Fig. 5).
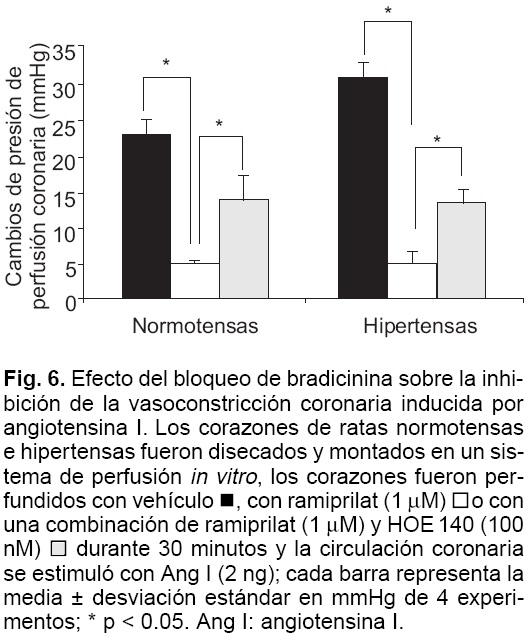
El efecto inhibitorio del ramiprilat sobre la vasoconstricción inducida por Ang I disminuyó en forma significativa cuando se bloquearon los receptores de Bk. La preincubación del corazón con ramiprilat (1 µM) inhibió el aumento de la presión de perfusión coronaria dependiente de Ang I en un 78% en ratas control y 82% en ratas hipertensas (p < 0.05). Cuando se coincubó la preparación con ramiprilat (1 µM) y HOE 140 (100 nM) esta inhibición fue de tan sólo 37% en corazones control y 55% en corazones hipertensos (Fig. 6).
Discusión
Nuestros datos confirman el efecto vasoconstrictor de Ang II en la circulación coronaria de rata, previamente descrito.15 Adicionalmente se demuestra que en el lecho coronario de animales hipertensos el efecto vasoconstrictor de Ang II es potenciado. La respuesta vasoconstrictora más intensa en los corazones de ratas hipertensas sugiere que durante el desarrollo de esta patología hay cambios a este nivel que hacen más eficiente la conversión de Ang I en Ang II, lo cual es compatible con evidencias experimentales en otros modelos de hipertensión arterial sistémica (HAS).16 Sin embargo, es importante señalar que estas observaciones se basan en evidencias farmacológicas, comparando el porcentaje de inhibición de la vasoconstricción coronaria generado por un inhibidor de ECA en los diferentes grupos experimentales, lo cual sugiere la necesidad de medir directamente la concentración de Ang en los lechos coronarios, con el fin de determinar exactamente la contribución de ECA en la circulación coronaria de ratas normotensas comparado con ratas hipertensas.
La vasoconstricción coronaria generada al administrar Ang I al sistema de perfusión confirma que a nivel de circulación coronaria de ratas normotensas e hipertensas hay conversión de Ang I en Ang II, lo cual es congruente con datos experimentales en otros modelos animales como puercos,17 perros18 y hámsters.19
Se han descrito variaciones entre especies en cuanto a la vía predominante de conversión de Ang I en Ang II en el corazón.5 En humano, perro y hamster se ha documentado la presencia de quimasa, la cual convierte Ang I en Ang II.7,18,19 Estudios en corazones de humano y perro muestran que quimasa es responsable de más del 90% de la formación de Ang II en estractos tisulares,20-22 mientras que ECA es la encargada de la conversión de más del 80% de Ang II en el lecho coronario.19,23,24 Se sugiere que hay diferentes compartimientos a nivel cardiaco, los cuales presentan variaciones en las vías enzimáticas encargadas de la conversión de Ang I en Ang II, que pueden cambiar en presencia de patología.18
En rata se propone que la generación de Ang II depende prácticamente de ECA,5,15 pero existe la posibilidad de que vías de conversión de Ang, alternas a ECA, sean importantes en estados patológicos.15 En este trabajo, la conversión de Ang I en Ang II en la circulación coronaria de rata pudiera estar mediada prioritariamente por ECA, puesto que el bloqueo agudo de esta enzima mediante preincubación con ramiprilat in vitro prácticamente abolió la vasoconstricción inducida por Ang I en corazones de ratas normotensas e hipertensas. Sin embargo, la medición de las concentraciones de Ang son indispensables para determinar con certeza el nivel de síntesis de Ang II en este grupo de animales. Aún más, al estar evaluando cambios en la circulación coronaria, no pudimos descartar que en el tejido cardiaco exista un componente tisular independiente de ECA.
Con el objeto de demostrar si el bloqueo de ECA in vivo modifica el efecto vasoconstrictor de Ang en la circulación coronaria, se valoró el efecto del tratamiento con ramipril vía oral durante 7 días sobre la conversión de Ang I en Ang II a nivel coronario. La inhibición de ECA in vivo mediante el tratamiento con ramipril vía oral, potenció la vasoconstricción inducida por Ang I tanto en ratas normotensas como en las hipertensas y además disminuyó el efecto del ramiprilat in vitro en ambos grupos. Esto pudiera deberse a que el bloqueo prolongado de ECA genera un fenómeno de escape, en el cual se inducen cambios en la actividad y/o expresión de ECA o se favorece la presencia de otras vías enzimáticas que generan Ang II. En rata se ha observado que la quimasa no hidroliza Ang I para formar Ang II, sino que favorece la degradación de Ang II,25 por lo que las vías enzimáticas independientes de ECA probablemente son de otra naturaleza.
Al bloqueo de ECA se le han atribuido acciones locales adicionales, no relacionadas a la reducción en la presión arterial, tales como la regresión de la hipertrofia del músculo cardiaco con reducción de la masa cardiaca además de mejorar la función hemodinámica de la circulación coronaria, y que pueden ser mediadas por Bk.10,26 Además se ha observado en algunos modelos animales, que las quininas endógenas contribuyen sustancialmente a la respuesta vascular renal ante la presencia de inhibición de ECA, sin embargo parece existir una variación importante inter-especie.5 En circulación coronaria de rata se ha reportado que el metabolismo de Bk depende predominantemente de ECA.13 Nuestros datos experimentales sugieren que el efecto de ramiprilat sobre la vasculatura coronaria de rata pudiera estar mediado parcialmente por Bk, ya que el bloqueo de la vasoconstricción inducida por Ang I se revirtió parcialmente cuando se inhibió el efecto vasodilatador de Bk. La inhibición del metabolismo de Bk pudiera ser un componente terapéutico importante del bloqueo de ECA.
Conclusiones
La conversión de Ang I en Ang II en el corazón de rata está mediado prioritariamente por ECA en animales normotensos e hipertensos. Sin embargo, la inhibición de ECA in vivo pudiera estar involucrada en cambios de actividad y/o expresión de ECA y en la inducción de otras vías enzimáticas que generan Ang II. Aún más, es posible que la actividad de Bk determine parcialmente el efecto de la inhibición de ECA por ramiprilat a nivel coronario, lo cual abre la posibilidad de que en proyectos futuros se puedan establecer con certeza los mecanismos por los cuales Bk participa en la regulación del tono coronario, en condiciones de inhibición de la ECA.
Referencias
1. DOSTAL DE, BAKER KM: The cardiac renin-angiotensin system. Conceptual, or a regulator of cardiac function? Circ Res 1999; 85: 643-650. [ Links ]
2. MAASSEN VANDENBRINK A, DE VRIES R, SAXENA PR, SHALEKAMP MA, DANSER AH: Vasoconstriction by in situ formed angiotensin II: role of ACE and chymase. Cardiovasc Res 1999; 44(2): 407-415. [ Links ]
3. NUSSBERGER J, BRUNNER DB, WAEBER B, BRUNNER HR: Specific measurement of angiotensin metabolites and in vitro generated angiotensin II in plasma. Hypertension 1986; 8: 476-482. [ Links ]
4. MENTO PF, WILKES BM: Plasma angiotensins and blood pressure during converting enzyme inhibition. Hypertension 1987; 9(6 Pt 2): III42-III48. [ Links ]
5. HOLLENBERG NK: Implications of species difference for clinical investigation. Studies on the reninangiotensin system. Hypertension 2000; 35(part 2): 150-154. [ Links ]
6. DZAU V, SASAMURA H, HEIN L: Heterogeneity of angiotensin synthetic pathways and receptor subtypes: physiological and pharmacological implications. J Hypertens 1993; 11(suppl 3): S13-S18. [ Links ]
7. URATA H, KINOSHITA A, MISONO KS, BUMPUS FM, HUSAIN A: Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem 1990; 265(36): 22348-22357. [ Links ]
8. AKASU M, URATA H, KINOSHITA A, SASAGURI M, IDEISHI M, ARAKAWA K: Differences in tissue angiotensin II-forming pathways by species and organs in vitro. Hypertension 1998; 32(3): 514-20. [ Links ]
9. LIAO Y, HUSAIN A: The chymase-angiotensin system in humans: biochemistry, molecular biology and potential role in cardiovascular diseases. Can J Cardiol 1995; 11(Suppl F): 13F-19F. [ Links ]
10. LINZ W, SCHÖLKENS BA: A specific B2-bradykinin receptor antagonist HOE140 abolishes the antihypertrophic effect of ramipril. Br J Pharmacol 1992; 105(4): 771-772. [ Links ]
11. PITT B: The anti-ischemic potential of angiotensin-converting enzyme inhibition: insights from the heart outcomes prevention evaluation trial. Clin Cardiol 2000; 23(7 Suppl 4): IV9-14. [ Links ]
12. ERDÖS EG: Some old and some new ideas on kinin metabolism. J Cardiovasc Pharmacol 1990; 15 (Suppl 6): S20-S24. [ Links ]
13. DUMOULIN MJ, ADAM A, BLAIS C JR, LAMONTAGNE D: Metabolism of bradykinin by the rat coronary vascular bed. Cardiovasc Res 1998; 38: 229-236. [ Links ]
14. CARRETERO OA, SCICLI AG: Local hormonal factors (intracrine, autocrine and paracrine) in hypertension. Hypertension 1991; 18(Suppl 3): I58-I69. [ Links ]
15. MÜLLER DN, FISCHLI W, CLOZEL JP, HILGERS KF, BOHLENDER J, MÉNARD J, ET AL: Local angiotensin II generation in the rat heart. Role of renin uptake. Circ Res 1998; 82: 13-20. [ Links ]
16. FERNÁNDEZ-ALFONSO MS, GONZÁLEZ C: Nitric oxide and the renin-angiotensin system. Is there a physiological interplay between the systems? J Hypertens 1999; 17: 1355-1361. [ Links ]
17. VAN KATS JP, DANSER AHJ, VAN MEEGEN JR, SASSEN LMA, VERDOUW PD, SCHALEKAMP MA: Angiotensin production by the heart. A quantitative study in pigs with the use of radiolabeled angiotensin infusions. Circulation 1998; 98: 73-81. [ Links ]
18. WEI C, MENG QC, PALMER R, HAGEMAN GR, DURAND J, BRADLEY WE, ET AL: Evidence for angiotensin-converting enzyme and chymase-mediated angiotensin II formation in the interstitial fluid space of the dog heart in vivo. Circulation 1999; 99: 2583-2589. [ Links ]
19. NISHIMURA H, BUIKEMA H, BALTATU O, GANTEN D, URATA H: Functional evidence for alternative ang II-forming pathways in hamster cardiovascular system. Am J Physiol 1998; 275: H1307-H1312. [ Links ]
20. BALCELLS E, MENG QC, JOHNSON WH JR, OPARIL S, DELL'ITALIA LJ: Angiotensin II formation from ACE and chymase in human and animal hearts: methods and species considerations. Am J Physiol 1997; 273: H1769-H1774. [ Links ]
21. WOLNY A, CLOZEL J, REIN J, MORY P, VOGT P, TURINO M, ET AL: Functional and biochemical analysis of angiotensin II-forming pathways in the human heart. Circ Res 1997; 80: 219-227. [ Links ]
22. URATA H, HEALY B, STEWART RW, BUMPUS FM, HUSAIN A: Angiotensin II-forming pathways in normal and failing human hearts. Circ Res 1990; 66: 883-890. [ Links ]
23. ZISMAN LS, ABRAHAM WT, MEIXELL GE, VAMVAKIAS BN, QUAIFE RA, LOWES BD, ET AL: Angiotensin II formation in the intact human heart. Predominance of the angiotensin-converting enzyme pathway. J Clin Invest 1995; 96(3): 1490-1498. [ Links ]
24. BALCELLS E, MENG QC, HAGEMAN GR, PALMER RW, DURAND JN, DELL'ITALIA LJ: Angiotensin II formation in dog heart is mediated by different pathways in vivo and in vitro. Am J Physiol 1996; 271: H417-H421. [ Links ]
25. LE TRONG H, NEURATH H, WOODBURY RG: Substrate specificity of the chymotrypsin-like protease in secretory granules isolated from rat mast cells. Proc Natl Acad Sci USA 1987; 84: 364-367. [ Links ]
26. NUNEZ E, HOSOYA K, SUSIC D, FROHLICH ED: Enalapril and losartan reduced cardiac mass and improved coronary hemodynamics in SHR. Hypertension 1997; 29(1 Pt 2): 519-24. [ Links ]

