











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
En los años 30, McCune, y un año más tarde, Albright, presentaron pacientes con ciertas características: displasia fibrosa de uno o varios huesos, manchas de color café con leche en el cuerpo y pubertad precoz.1,2 Posteriormente, se identificó a estos enfermos como portadores del síndrome que lleva sus nombres. De un modo progresivo, se fueron agregando otras condiciones como exceso de hormona del crecimiento, eliminación renal anormal de fosfato con o sin raquitismo/osteomalacia, síndrome de Cushing y, rara vez, la participación de otros órganos y sistemas como el cerebro, ojos, dientes, hígado, corazón, paratiroides o páncreas.
Este síndrome es una condición rara y no hereditaria. En nuestro país, sólo hemos encontrado tres comunicaciones al respecto.3-5 Aunque no se cuenta con datos de prevalencia disponibles, se ha comentado que puede ocurrir en rangos que van de 1/100,000 a 1/1,000,000. Los estudios genéticos de las llamadas proteínas G, relacionadas con el desarrollo de los tejidos musculoesqueléticos y hormonales, han permitido identificar que las alteraciones genéticas están en el brazo largo del cromosoma 20 (20q13.2), en el gen denominado GNAS1. Existe mutación en mosaicismo, al azar en todas o sólo algunas células. Además, no es hereditaria y los pacientes sufren displasia fibrosa, causa de malformaciones óseas en diversos sitios.6,7
Nuestro objetivo es presentar los hallazgos clínicos de un adolescente que acudió a consulta por una lesión en la planta de un pie y tenía manifestaciones de este síndrome.
Caso clínico
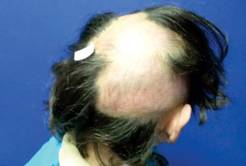
Un joven de 16 años se presentó a consulta acompañado por un familiar, por una úlcera de unos ocho meses de evolución en la parte posterior de la planta del pie derecho, dolorosa y que no había cedido a cuidados generales. Desde niño tuvo la extremidad inferior derecha más corta y delgada que la izquierda, lo que motivó una marcha irregular. Desde que nació tenía manchas en el cuerpo, que también se extendían a ambas piernas. Por varios años, había notado que en casi tres cuartas partes de la cabeza no tenía cabello, por lo que todo el tiempo usaba una gorra.
A la exploración, se encontró a un joven en buenas condiciones generales, con gorra puesta. Peso: 61 kg, estatura: 1.65 m. Tensión arterial, normal. La marcha, difícil por apoyar la parte delantera del pie derecho e inclinar el tronco hacia afuera. El tórax derecho, cubierto por piel con una mancha color café que se extendía hasta el pie y hacia el brazo. En el lado izquierdo también había mancha, pero en menor proporción, aunque distalmente se extendía hasta el talón. No se apreciaron anomalías a la auscultación del tórax o al examen del abdomen. La extremidad inferior derecha era más delgada y corta que la izquierda. En la planta del pie derecho tenía una úlcera de 11 x 6 cm, localizada en la mitad posterior del pie, con bordes eritematosos y evidencia de sangrado superficial. No se apreciaron huesos o tendones expuestos. Los pulsos periféricos eran normales. Durante el examen se apreció un crecimiento de los genitales externos; el paciente no refirió dolor. Al retirar la gorra se apreció una zona de alopecia que abarcaba las dos terceras partes del cuero cabelludo (Figuras 1, 2, 3, 4 y 5).
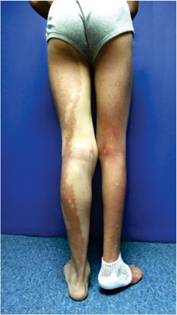
Figura 1: Vista posterior de las extremidades inferiores. La mancha café se extiende hasta los pies.
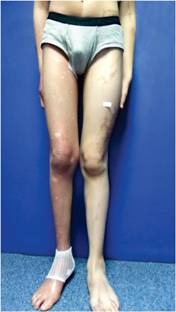
Figura 2: Acortamiento y adelgazamiento del miembro inferior derecho, con manchas hasta el pie. En el lado izquierdo se aprecian algunas manchas.
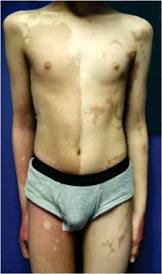
Figura 3: Manchas café con leche de predominio en la mitad derecha del tórax y abdomen, y que se extienden hacia el muslo. En el lado izquierdo también existen manchas. Se aprecia un aumento del tamaño de los genitales externos.
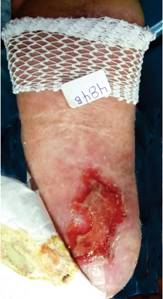
Se le efectuó aseo de la úlcera y se le colocó un vendaje acolchonado suave. Se le dieron indicaciones para el cuidado de la úlcera, la colocación de vendaje suave y la obtención de una plantilla especial para evitar la presión en esa zona. Se le dio una nueva cita, a la que no acudió. Cuando la asistente llamó al teléfono que habían dejado, informaron que ya se habían cambiado de domicilio e ignoraban la nueva dirección.
Discusión
De una manera clásica, se define al síndrome de McCune-Albright por la tríada clínica de displasia fibrosa de hueso, manchas café con leche en el cuerpo y pubertad precoz.6 Las alteraciones óseas pueden afectar uno o varios huesos y presentarse con dolor o trastornos para caminar, con o sin fractura. Tiene cierto predominio en pacientes del sexo femenino, en quienes se manifiesta por sangrado vaginal o crecimiento de mamas; en los hombres puede haber crecimiento genital y conducta sexual precoz. Si hay otras endocrinopatías, puede haber manifestaciones tiroideas, en el crecimiento -como un síndrome de Cushing- y eliminación anormal por vía renal de fosfato, entre otros.7 En la cabeza pueden existir manifestaciones oftálmicas, auditivas o esqueléticas de severidad variable. Salenave y sus colaboradores revisaron 112 pacientes y señalaron que la acromegalia está presente en el 20 a 30% de quienes padecen de este síndrome y tienen las características particulares de diagnóstico y tratamiento.8 Hasta donde sabemos, éste es el primer informe de alopecia areata asociada a este síndrome.
El tratamiento es muy variado, depende de la edad del paciente y los órganos involucrados, pero es indudable que se requiere de la participación de un equipo con múltiples especialistas, pediatras, radiólogos, internistas, cirujanos, ortopedistas, genetistas, dermatólogos, oftalmólogos, endocrinólogos y otros, según el caso. En cada especialidad, el médico asignado será el encargado de otorgar el abordaje necesario.9 Así, aunque no hay tratamiento para la displasia fibrosa de los huesos, el ortopedista se enfocará en optimizar la función y minimizar la morbilidad debida a fracturas y deformidades. Para atender la precocidad sexual, el ginecólogo puede usar el letrozole y un receptor como el tamoxifén; en hombres jóvenes, las opciones no están establecidas. En un estudio de 54 varones con síndrome de McCune-Albright, Boyce y su grupo encontraron como hallazgo histopatológico predominante la hiperplasia de las células de Leydig.10
El metimazole mejora las condiciones de hipertiroidismo persistente, aunque se puede requerir la tiroidectomía. Para mejorar el exceso de hormona del crecimiento, se puede recurrir al octreótido y a un antagonista del receptor de la hormona del crecimiento, como el pegvisomant. Se deben hacer estudios de la columna vertebral para descartar escoliosis progresiva. Es recomendable evitar deportes y actividades que puedan comprometer a los huesos.
En hombres, se deben evaluar las características genitales y vigilar con ultrasonido testicular, con la participación activa de urólogos interesados. Es útil vigilar los niveles de fósforo para observar la hipofosfatemia. Casi cada año se debe obtener evaluación ocular y otológica y tomografía del cráneo para vigilar el estado de displasia ósea en esa zona, con afección funcional. En ocasiones, es necesario descomprimir alguna presión sobre el nervio óptico.11 Puede requerirse la participación de cirujanos reconstructivos y odontólogos capacitados para corregir deformaciones orofaciales. Pichard y sus colegas informaron de cuatro pacientes con este síndrome que, además, tenían pigmentación de la mucosa oral en la infancia o juventud.12
En el caso aquí presentado, sus condiciones socioeconómicas evitaron que regresara a consulta para continuar su estudio y tratamiento, pero fue posible integrar el síndrome de McCune-Albright con las características clínicas.6,12,13

