










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.48 no.4 Ciudad de México oct./dic. 2004
Investigación
Effect of Formulation Variables on Verapamil Hydrochloride Release from Hydrated HPMC Matrices
Marcela Ramírez-Campos y Leopoldo Villafuerte-Robles1
Departamento de Farmacia de la Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional de México. Carpio y Plan de Ayala s/n, Col. Santo Tomás, C. P. 11340 D. F. México. Fax: (52) 5396 3503. E-mail: lvillaro@encb.ipn.mx
Recibido el 16 de abril del 2003.
Aceptado el 10 de diciembre del 2004.
Abstract
Among different variables that influence drug release from hydrated hydrophilic matrices, the polymer proportion, the drug dose and the matrix pH can be included. In a first approach to formulate a transdermal drug delivery system, these variables have been used to modify the drug release rate and to examine its effect on the release mechanism. Hydrated matrices were prepared varying the matrix proportion of hydroxypropyl methylcellulose (HPMC), the pH and the amount of verapamil hydrochloride loaded. The matrices release behavior (USP 23 apparatus 5, paddle over disk method, at 150 rpm) was examined using 900 mL of an aqueous solution of NaCl (0.9%) as dissolution medium. The increase of the HPMC matrix proportion reduced the release rate of the drug. The release profiles showed zero order kinetics for drug dissolution proportions up to 70%. The increase of the matrix drug load increases slightly the release rate. The increase of the matrix pH from 5.0 to 8.0 increased the release rate. The effect of both pH and drug load becomes lesser as the matrix polymer proportion increases from 5% to 15% w/w. The use of a Collodion membrane covering the hydrated matrices decreases two orders of magnitude the release rate of verapamil hydrochloride.
Key words: Sustained release, verapamil, transdermal delivery, HPMC, pH.
Resumen
Entre las variables de drogas que tienen influencia sobre la velocidad de liberación desde matrices hidrofílicas hidratadas, se encuentran la proporción del polímero, la dosis del fármaco y el pH de la matriz. En una primera aproximación a la formulación de un sistema de liberación transdérmico de fármacos, se utilizan estas variables para modificar la velocidad de liberación y para estudiar su efecto sobre el mecanismo de liberación. Las matrices hidratadas se prepararon variando la proporción de la hidroxipropilmetilcelulosa, el pH y la cantidad cargada del clorhidrato de verapamil en la matriz. Los estudios de liberación desde las matrices se llevaron a cabo utilizando 900 mL de una solución acuosa de NaCl (0.9%) como medio de disolución. El incremento de la proporción de HPMC en la matriz redujo la velocidad de liberación del fármaco. Los perfiles de liberación mostraron una cinética de orden cero para proporciones del fármaco disuelto de hasta 70%. El aumento de la carga del fármaco en la matriz aumentó ligeramente la velocidad de liberación. El aumento del pH de la matriz de 5.0 hasta 8.0 aumentó la velocidad de liberación. El efecto conjunto del pH y de la dosis de carga del fármaco disminuye conforme la proporción del polímero en la matriz aumenta de 5% a 15% p/p. La utilización de una membrana de Colodión para cubrir las matrices hidratadas disminuye en dos órdenes de magnitud la velocidad de liberación del clorhidrato de verapamil.
Palabras clave: Liberación sostenida, verapamil, liberación transdérmica, HPMC, pH.
1 Introduction
The potential of using the intact skin as the port for continuous transdermal drug delivery beyond the boundary of topical medication has been recognized. Transdermal delivery systems are classified in different categories, according to the technological basis of their approach, including the membrane permeation-controlled and the matrix diffusion-controlled transdermal therapeutic systems [1].
The transdermal permeation can be analyzed as a diffusion process through a passive membrane. The concentration of the applied solute species at the surface layers is not usually equal to but related to its concentration in the applied vehicle in accord with the vehicle/stratum corneum sorption isotherm. The linear isotherm can be defined in terms of the distribution coefficient between the vehicle and the stratum corneum. It is considered that the transdermal flux is directly proportional to the concentration difference across the skin barrier [2].
Verapamil belongs to the group of calcium channel antagonists used in the management of essential hypertension. It is mostly used in a conventional tablet form from a minimal dose of 40 mg to a maximal dose of 180 mg. In a slow release tablet, the doses are between 120 and 240 mg. About 80% of the orally applied dose undergoes a first pass effect, mainly in the liver [3]. The preparation of floating pellets has been used to get a more effective release and absorption of verapamil due to a higher solubility of the drug in the stomach [3]. Another attempt to avoid problems of verapamil solubility by oral administration is the use of organic acids to maintain the pH low and the addition of enteric materials to open pores in a matrix at the pH of the intestine [4]. However, the still low bioavailability associated with the oral route can be avoided by transdermal administration [5].
Verapamil has a short half-life and requires frequent dosing. The permeability characteristics of verapamil show poor skin permeation properties; the permeation is dependent on the drug concentration and pH of the donor solution [6].
Verapamil patches showed zero-order release from patches made of different hydrophilic and hydrophobic polymers. It has been considered that patches made of mixtures of Eudragit RL100 and hydroxypropyl methylcellulose are suitable for transdermal delivery. This type of verapamil administration avoids the first pass effect and produces extended release [7].
Polymer matrices make good reservoirs for verapamil sustained release medications. There are many hydrophilic polymers used for transdermal drug delivery systems like polyvinyl alcohol and polyvynilpyrrolidone [5] and hydroxypropyl methylcellulose [8]. Different formulations based on polyvinyl alcohol and polyvynilpyrrolidone showed release profiles with an almost zero order release kinetics.
Factors that affect the transdermal permeation of drugs, includes pH, drug concentration and partition coefficient. Studies using as donor a saturated solution of verapamil, at pH of 6.15, showed that the permeation rate through hairless mouse skin is approximately proportional to verapamil concentration in the donor solution. Verapamil is a weak base, consequently, as the pH decreases, the total solubility (ionized + nonionized) increases. However, the intrinsic solubility (non-ionized drug) remains constant. The effect of pH of the donor solution on the permeation rate of verapamil show maximal permeation rates at pH 6-7. The nonionized form of verapamil, showed in the following, was found to have a higher permeability to hairless mouse skin from aqueous buffer solutions [9].
Drug permeation through the skin following transdermal delivery is also influenced by various design factors such as polymer species, internal structure of the polymer matrix and drug-loading dose [10].
The effects of the above-mentioned factors must be elucidated for developing an optimum delivery system, first in vitro and then in vivo. In the present study, we have verified the effects of polymer concentration, drug loading dose and pH as well as their combinations on the in vitro release profile of verapamil hydrochloride from hydrated matrices of hydroxypropyl methylcellulose.
2 Materials and methods
2.1. Materials
The pharmaceutical excipient hydroxypropyl methylcellulose 4000 (HPMC), Methocel, obtained from Dow Chemicals and the drug verapamil hydrochloride (VER), obtained from Química Knoll de México and Kener de México, were used as received. The Collodion membrane was obtained from Sigma with a molecular weight cut-off of 12 000 Da.
2.2. Methods
2.2.1. Matrix preparation
Corresponding quantities of HPMC were weighed to get concentrations of 5, 10 and 15% in buffer solutions. The polymer was dissolved mixing manually until a homogeneous gel was obtained. The hydrated HPMC matrices (1.3 g) were loaded with different quantities of verapamil hydrochloride (300, 480 and 1000 mg) by manual blending in a mortar until a homogenous dispersion was obtained. The matrices with a given pH were prepared with a buffer solution pH 5.0 (sodium acetate/acetic acid 0.1M), pH 6.0 and pH 8.0 (potassium phosphate/NaOH 0.2M), before the drug was loaded.
2.2.2. Dissolution methodology
Dissolution studies were carried out at 32ºC and 150 rpm, with the USP 23 [11] dissolution apparatus 5 (paddle over disk method) in 900 mL of an aqueous solution of NaCl (0.9%). The samples were placed in a stainless steel assembly designed to hold the hydrated matrices. This assembly is circular with a diameter of 25.4 mm and 3 mm and 5 mm depth. An especially designed stainless steel ring is used to fix a stainless steel disk with multiple perforations (Millipore) or a Collodion membrane, to cover and protect the sample.
Samples were withdrawn at predetermined time intervals, filtered, diluted with HCl 0.1N and analyzed spectrophotometrically at a wavelength of 230 nm (Beckman DU-650 spectrophotometer). Dissolution studies were performed with three individual samples of each different formula. The results for each time point of the three different dissolution curves obtained are registered as an average in the figures.
3 Results and discussion
3.1. Effect of verapamil hydrochloride load on the release profile of hydrated matrices
The assessment of the release properties of transdermal therapeutic systems represents an important test for its pharmaceutical performance. For this test, samples are placed in direct contact with an aqueous receptor medium or inserting a permeable membrane in between [12].
Figure 1 shows the release profile of verapamil hydrochloride from hydrated matrices containing 5% HPMC at pH 5.0. In this case, the release profiles correspond to samples covered with the stainless steel disk with multiple perforations, to fix and protect the sample. The dissolution conditions are adequate for a fast drug release. The time necessary to release the total quantity of the drug loaded is dependent of the matrix drug content. An increasing drug load of the matrix increases correspondently the time necessary for an entire release of the drug. The release rate determined as the slope of the curves shows a similar magnitude for all different matrices at the beginning of the dissolution process. The release rate of verapamil hydrochloride increases slightly with increasing drug loads with time.
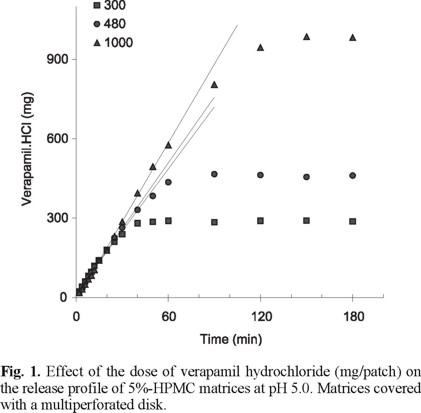
The dissolution rate of matrices containing different drug contents remains practically constant most of the time. The release kinetics is considered of zero order for the most part of the drug dissolution. Under the given rapid dissolution conditions, it seems that the matrices reach in the early stages a steady state for the drug transport independent of the drug loaded, changing slightly with time. The transport of the drug through the matrix is considered a combination of diffusion and relaxation/erosion of the matrix.
The diffusion coefficient most likely increases gradually during the swelling process as the polymer chains are diluted with imbibed solvent. The result is a reduction of the local viscosity [13]. The extreme case of matrix relaxation is the matrix erosion.
There are evident problems when a matrix intended to be a patch either releases the drug very quickly, with a rate faster than the biological membrane penetration, or undergoes dissolution in the receptor solution. To avoid this, a membrane can be used to restrict the drug transport and to protect the matrix during the dissolution testing.
The release profile of matrices with the same formula than those above-mentioned but covered with a hydrated Collodion membrane instead of the stainless steel disk shows two orders of magnitude smaller release rates.
It seems that the transport through the Collodion membrane is the slowest step of the transport of the drug and determines the matrices drug release rate. However, and given the molecular weight cut of the membrane, the reduction of the release rate can also be attributed to a much smaller erosion of the polymeric matrix. The release kinetics followed up to 300 min is considered of zero order. The drug transport could be also a combination of diffusion through the matrix, matrix relaxation due to water absorption and a little erosion.
Matrix relaxation becomes more important as water can flow through the membrane to alter the hydration state of the hydrophilic matrix. This may be a particular problem for occlusive patches applied to the skin over extended periods [12]. The same problem can be observed by the dissolution test of patches made of hydrophilic matrices.
3.2. Effect produced by the polymer concentration on the drug release profile
Literature review indicates that there was a decrease in the drug release as the concentration of polymer increased when verapamil hydrochloride was formulated as transdermal patch using sodium carboxymethyl guar as polymer matrix [7].
For the penetration of a small molecule in polymers, it is required cooperative movements of several monomeric units of the polymer chain. The mobility of the polymer chains thus controls the rate of diffusion, and factors affecting the chain mobility will influence the diffusion coefficient. One of the key factors is the concentration. The diffusion coefficient decreases as the concentration of the polymer molecules increases. As the polymer molecules concentration increases, they begin to interact with each other hydrodynamically, leading to a concentration dependence of the diffusion coefficient. The interaction of polymer molecules is characterized by the overlap of the polymer chains and intermolecular entanglement leading to a dynamic network structure [14].
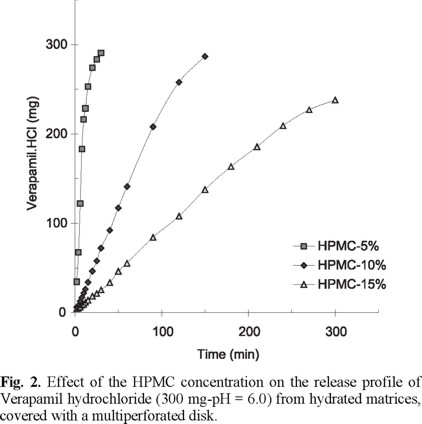
The matrix increase of HPMC affects substantially the release process. The release profiles of verapamil hydrochloride matrices show decreasing release rates with an increasing HPMC content (figure 2). The release mechanism of matrices shown in figure 2 is considered as zero order for a verapamil hydrochloride release up to about 70%. The results are attributed to an increased interaction of polymer molecules leading to an increased viscosity of the polymeric matrix as the HPMC proportion increases.
3.3. Effect of pH of the polymeric matrix on the release profile
The effect of pH of the hydrated matrix containing verapamil hydrochloride was studied at three pH levels 5.0, 6.0 and 8.0. Figure 3 shows the release profile of verapamil hydrochloride (480 mg) from HPMC hydrated matrices containing 5% of this polymer. There is no lag time in the release profiles showed in figure 3 and some other because of the hydrated state of the polymeric matrices prior the dissolution test begins. As it can be seen, the release rate of verapamil hydrochloride increases as the pH increases. At pH 5.0, the release rate, calculated as the slope of the curve, was 8.88 mg/min; at pH 6.0 the release rate was 22.8 mg/min while at pH 8.0 it was 38.6 mg/min. These and all other calculated release rates, expressed as mg/min, are summarized in table 1. An increasing release rate was observed as the pH increases in spite of a resultant increase in the base form of verapamil. Verapamil is a weak base with a pKa of about 8.7. Consequently, as the pH of the hydrated matrix decreases the total solubility of the ionized plus the nonionized forms should increase. However, the intrinsic solubility of the base remains constant [9].
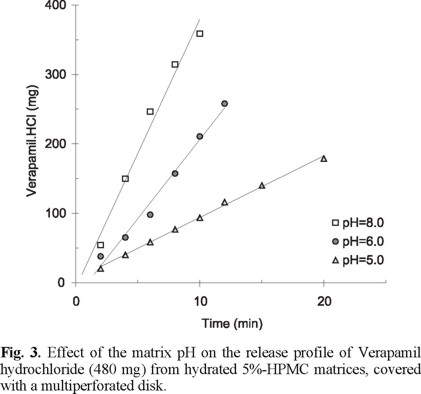

The increase in dissolution rate of verapamil hydrochloride as the pH increases can be attributed to a change in the hydration properties of the gel because of the ions of the buffer. It seems that the added salts enhance the verapamil hydrochloride dissolution. Some substances like propanolol have been found to increase the hydration of HPMC while many electrolytes depress the gel points of the polymers by affecting dehydration [15].
Another possibility for higher release rates with increasing values of pH could be a reduction of the hydrodynamic radius, as verapamil hydrochloride becomes a molecule with lower polarity. The verapamil hydrochloride molecules carry a lesser amount of positive or negative electric charges. The molecule transport takes place through interstices of polymer chains filled with aqueous medium and is dependent of the molecule radius. However, the radius is not that of the bare molecule but that of the hydrodynamic particle. The hydrodynamic particle consists of the diffusant molecule and any solute or solvent molecules bound or adsorbed to the surface or within the diffusant. Therefore, hydration of the diffusant increases the hydrodynamic radius [16].
3.4. Effect of combined variables on the release rate of verapamil hydrochloride from HPMC matrices
Figure 4 shows the effect of pH and the polymer proportion on the release rate of verapamil hydrochloride from hydrated matrices. The response surface was calculated utilizing the average values of all different curves obtained from matrices containing different drug load, for each given pH value and HPMC concentration. The effect of pH is quite small at HPMC concentrations of 15%, increasing the pH effect as the HPMC concentrations decreases. In the same way, as the pH increases it gives emphasis to the effect of different proportions of HPMC.
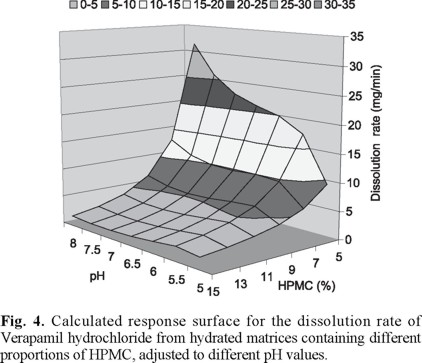
It seems that the lower the release rate produced by a first variable the smaller the effect of a second variable. The maximal effect of the second variable on the release rate is observed at the lowest effect of the first variable. When the polymer proportion increases, it reduces heavily the release rate of verapamil hydrochloride and the pH effect on the release rate is practically zero. On the other hand, although at pH 5.0 the effect of the polymer concentration does not disappear, it is reduced. In the main, the effect of the polymer concentration prevails over the pH effect.
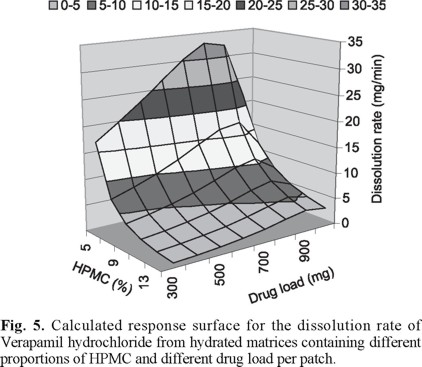
The combined effect of polymer proportion and verapamil hydrochloride dose is showed in figure 5. The response surface was calculated utilizing the average values of all different curves obtained from matrices adjusted at different values of pH, for each given HPMC concentration and drug load. In the same way as in figure 4, increasing proportions of the polymer lesser or hide the effect of the verapamil hydrochloride load. The effect of the amount of verapamil hydrochloride loaded becomes more important as the matrix polymer proportion decreases. Increasing polymer proportions in the matrix reduce the release rate. Increasing amounts of verapamil hydrochloride loaded in the matrix increases the release rate.
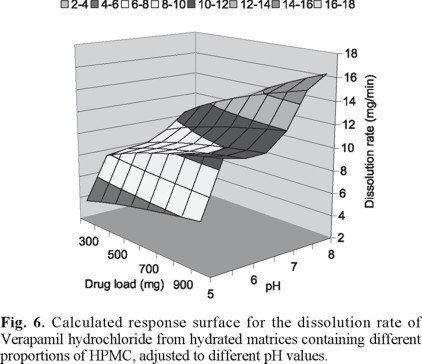
The combined effect of pH and verapamil hydrochloride dose is showed in figure 6. The response surface was calculated utilizing the average values of all different curves obtained from matrices adjusted at different HPMC proportions, for each given pH and drug load. Increasing matrix pH values increase the dissolution or release rate of the drug. The effect of the amount of verapamil hydrochloride loaded becomes more important as the matrix pH increases. The increase of both the matrix pH and the amount of verapamil hydrochloride loaded in the matrix increases the release rate.
4. Conclusion
The drug load, the pH of the matrix and the polymer concentration of the matrix affects the release profile of verapamil hydrochloride from hydrated HPMC matrices significantly. Matrices with different drug load show similar release rates at the early stages of the dissolution process that change slightly with time. This is considered a consequence of solid particles of the drug in the polymeric matrix that allows a diffusion steady state of the drug that maintains a constant release rate. The effect of increasing values of pH that increase the release rate of verapamil hydrochloride has to be reviewed. This effect could not be clearly assigned to a particular physical variable. The effect of increasing polymer proportions to decrease the drug release rate is considered the more important variable determining the drug release rate. High polymer concentrations such as 15% w/w can hide from sight the effect of the other two variables, which are visible only at low polymer concentrations such as 5%. The dissolution method paddle over the disk allows great matrix erosion. The use of a collodion membrane can avoid this erosion but reduce in a great extent the release rate, in spite of a membrane molecular cut-off of 12 000 Da.
References
1. Chien, Y. W. (Editor). Transdermal controlled systemic medication. Marcel Dekker, New York - USA. 1987, pp. 14-15. [ Links ]
2. Chien, Y. W. Novel Drug Delivery Systems. Marcel Dekker, New York - USA. 1982, pp 160-161. [ Links ]
3. Sawicki, W. Eur. J. Pharm. Biopharm. 2002, 53, 29-35. [ Links ]
4. Munday, D. L. Drug Dev. Ind. Pharm. 2003, 29, 575-583. [ Links ]
5. Jain, G.K.; Sharma, A.K.; Agrawal, S.S. Int. J. Pharm. 1996, 130, 169-177. [ Links ]
6. Ritschel, W. A.; Agrawala, P. Acta Pharm. Tech. 1988, 34, 156-159. [ Links ]
7. Devi, V. K.; Saisivam, S.; Maria, G. R.; Deepti, P. U. Drug Dev. Ind. Pharm. 2003, 29, 495-503. [ Links ]
8. El-Kattan, A. F.; Asbill, Ch. S.; Michniak, B. B. Int. J. Pharm. 2000, 198, 179-189. [ Links ]
9. Shah, H.S.; Tojo, K.; Chien, Y.W. Int. J. Pharm. 1992, 86, 167-173. [ Links ]
10. Tojo, K.; Chien, Y. W. J. Chem. Eng. Japan. 1989, 22, 689-693. [ Links ]
11. USP 23 NF 18. United States Pharmacopoeial Convention, Inc., Rockville, MD, USA. 1995. Pp. 1796-1797. [ Links ]
12. Iordanskii, A. L.; Feldstein, M. M.; Markin, V. S.; Hadgraft, J.; Plate, N. A. Eur. J. Pharm. Biopharm. 2000, 49, 287-293. [ Links ]
13. Schott, H. J. Pharm. Sci. 1992, 81, 467-470. [ Links ]
14. Kou, J. H. Transport in polymer systems, in Amidon, G. L., Lee, P. I., Topp, E. M. Transport processes in pharmaceutical systems. Marcel Dekker, New York, 2000, pp. 451-452. [ Links ]
15. Mitchell, K.; Ford, J. L.; Armstrong, D. J.; Elliot, P. N. C.; Hogan, J. E.; Rostron, C. Int. J. Pharm. 1993, 100, 165-173. [ Links ]
16. Nerurkar, M. J.; Duddu, S.; Grant, D. J. W.; Rytting, J. H. Properties of solids that affect transport, in Amidon, G. L., Lee, P. I., Topp, E. M. Transport processes in pharmaceutical systems. Marcel Dekker, New York, 2000, pp. 600-601. [ Links ]

