










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.48 no.4 Ciudad de México oct./dic. 2004
Investigación
Simultaneous Spectrophotometric Determination of Isoperoxisomicine A1 and Peroxisomicine A1 Using Partial Least Square Regression Type 1 (PLS-1)
Ma. de la Luz Salazar,1 Pedro Luis López de Alba,2 Alfredo Piñeyro López3, and Noemí Waksman de Torres1*
1 Departamento de Química Analítica de la Facultad de Medicina de la Universidad Autónoma de Nuevo León. Apdo. Postal 2316, Sucursal Tecnológico, 64841 Monterrey, N. L., México. E-mail: nwaksman@fm.uanl.mx
2 Instituto de Investigaciones Científicas, Universidad de Guanajuato.
3 Departamento de Farmacología y Toxicología de la Facultad de Medicina de la Universidad Autónoma de Nuevo León, Monterrey, Nuevo León, México.
Recibido el 26 de julio del 2004.
Aceptado el 10 de diciembre del 2004.
Abstract
Multivariate calibration with regression by partial least squares (PLS-1) is herein proposed as an alternative method for the spectrophotometric quantification of isoperoxisomicine A1 in peroxisomicine A1 batches. In order to minimize the optimal factors necessary to obtain the calibration matrix, different parameters were evaluated. The adequate selection of the spectral regions proved to be important on the number of factors. In order to simultaneously quantify both analytes, the spectral region between 252 and 340 nm was selected. Recoveries for isoPA1 and PA1 were 97% and 103% respectively. The developed method was applied to two batches of PA1; the amount of isoPA1 found, showed no significant differences with those found by means of 1H RMN.
Key words: PLS-1, spectrophotometry, peroxisomicine, isoperoxisomicine, multivariate calibration.
Resumen
La calibración multivariante de los mínimos cuadrados parciales tipo 1 (PLS-1) es propuesta para la determinación espectrofotométrica de isoperoxisomicina (IsoPA1) en lotes de peroxisomicina A1 (PA1). Se estudiaron diferentes parámetros para minimizar el número de factores óptimo para la obtención de la matriz de calibrado. Se observó que la selección adecuada de las regiones espectrales muestra un efecto importante sobre el número de factores. Para la determinación simultánea de los analitos en estudio, se seleccionó la región espectral de 252 a 340 nm. Los porcentajes de recuperación obtenidos para IsoPA1 y PA1 cuando se trabajó en esta región espectral fueron entre 97 y 103. El método desarrollado se aplicó a dos lotes de peroxisomicina A1, cuantificándose el contenido de IsoPA1 con resultados comparables a los obtenidos por RMN 1H.
Palabras clave: PLS-1, espectrofotometría, peroxisomicina, isoperoxisomicina, calibración multivariante.
Introduction
Peroxisomicine A1, (3,3'-dimetil-3,3',8,8',9,9'-hexahydroxy-3, 3',4,4'-tetrahydro-(7,10')-biantracen-1,1'-(2H,2'H) dione) (PA1, figure 1), is a coupled hydroxyanthracenone isolated from the seeds of plants belonging to Karwinskia genus (Figure 1) [1].
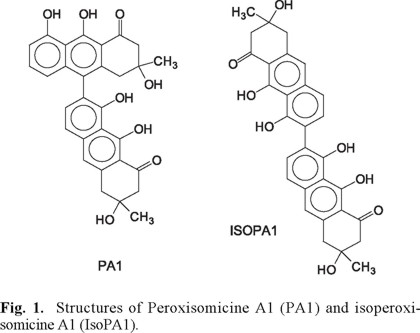
It displays a selective in vitro cytotoxicity for neoplastic cells derived from hepatic, pulmonary and colon tissues [2]; therefore PA1 has been patented as an anticancer agent [3] and is currently under preclinical studies. Thus, the extraction and purification procedures to obtain this compound in greater scale and as pure as possible have been improved in the last years. Despite all purification steps, final product batches of peroxisomicine A1 obtained in our laboratory contained between 3% and 5% of several isomers, being isoperoxisomicine A1 (IsoPA1, figure 1) the most persistent [4,5]. This is not surprising, as preparative HPLC (reverse phase) is the final purification step, and both compounds present similar retention times in most HPLC systems studied [6] .
Our goal is to develop analytical methods to quantify the contaminants present in PA1 batches.
Several biological and physicochemical assays are currently in use to carry out the quality control of PA1 batches. Physicochemical parameters in use are melting point, TLC (normal and reverse phase) and absorbance relationship at four different wavelengths, namely 220, 270, 415 and 430 nm.
More recently HPLC-DAD and 1H NMR methods have been proposed for assess the purity of PA1 [7,8]. Purity analysis using HPLC-DAD is accomplished through different methodologies, such as: normalized chromatogram matching, spectral matching and by absorbance ratio at 269, 280, 310 and 410 nm. However, although this analysis allowed to detect the presence of IsoPA1 in PA1 batches at concentrations as low as 1%, no direct correlation could be found between the IsoPA1 content and purity index [7]. Several chemometric methods have been proposed to deconvolute partially overlapped chromatographic peaks [9-12]. The Savistsky and Golay derivatization algorithm [13] has been used for this purpose with good results [7]; however, analytical signal originated from IsoPA1 (chromatographic peak) in PA1 batches is not sensible enough as to quantify with precision this compound by derivative spectroscopy. Multivariate calibration and neural networks have been used with good results to solve different analytical problems arising from highly overlapped analytical signals, even when the difference in the concentrations between the analytes is large [14-17].
In the present contribution we applied a multivariate calibration method with regression by partial least squares (PLS-1) to quantify up to 0.7% of IsoPA1 in PA1 batches.
Experimental
Instruments. A DU-7500 Beckmann Diode Array spectrophotometer was used (0.35 nm resolution). All mathematical data treatment operation of spectral data were carried out with GRAMS/32tmv.5 software package from Galactic Co., Salem, N.H., U.S.A.
Reagents. Peroxisomicine A1 and isoperoxisomicine A1 were isolated, purified and identified by NMR in our laboratory as previously described [8]. Solvents used were HPLC grade (Merck, Darmstadt). Water was purified by means of a Milli-Q system ( Millipore, Bedford, U.S.A.)
Procedure
Construction of the calibration model. The multivariate calibration analysis (PLS-1) was carried out by means of an experimental design for two components, namely: PA1 and IsoPA1 and 36 experiments. Mixture solutions were prepared in 5 mL volumetric flasks, using methanol as solvent. Concentration intervals were chosen from the lineal working ranges calculated for each compound: 0.4-10.0 mg—L-1 for IsoPA1 and 1-12 mg—L-1 for PA1. Absorption spectra of these samples were registered in the range of 200-600 nm against a blank of methanol. Spectra were transformed to ASCII format and further exported to PLS plus GRAMS 32 v-5 program. Quantification of the mixtures was accomplished by the partial least squares method in its type 1 algorithm version (PLS-1) [14, 18-20]. To select the minimum optimal factors, the minimum PRESS (Prediction Residual Error Sum of Squares) and F statistical analysis were employed.
Validation of the calibration model. The internal validation of the developed method, was settled down by the square of correlation coefficient calculation (R2, Pearson correlation coefficient) and the relative error of prediction (REP%). The external validation of the calibration model was accomplished with a set of 16 synthetic mixtures of random composition within the concentration range considered in the experimental design; predictive capacity for each component was estimated calculating the mean recovery percentage +/- standard deviation (%R ± SD), the SEC/SEP (standard prediction error) and the REP (relative prediction error) [16]. The statistical parameters were calculated according to the equations:
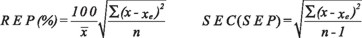
x is the real concentration, xe is the predicted concentration for one of the analytes in the samples (calibration or validation), is the mean concentration of the analyte in the same series and n is the number of samples evaluated.
Real samples analysis. The constructed experimental model was used to calculate the concentration of IsoPA1 in several PA1 batches. Samples were prepared dissolving 1 mg of PA1 in 1 mL of methanol. The concentrations calculated by means of this procedure were compared with the results obtained by the HPLC and 1H NMR methods already described [7,8].
Results and Discussion
Figure 2 shows the absorption spectrum for the individual component (PA1 and IsoPA1) in methanol. The great spectral similarity of both compounds and the low relative concentration of IsoPA1, makes difficult the quantification of the contaminant (IsoPA1) in PA1 batches, using zero-order or derivative UV-Vis spectra. Therefore, we decided to solve the analytical problem by the application of a mathematical algorithm, namely the calibration using partial least square regression type 1(PLS-1); the theoretical background of this method was previously published [14].
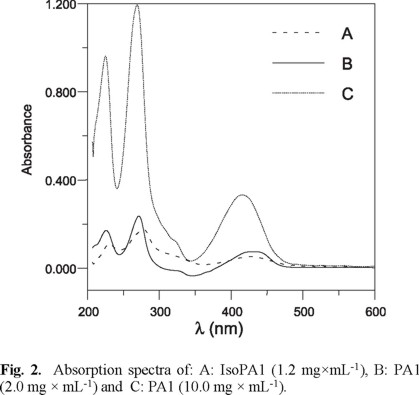
Calibration
With the aim of giving more weight to the analytical signal arising from the contaminant, which is the purpose of the present work, and in order to be able to quantify IsoPA1 with a high degree of confidence too, we decided to use a factorial design of type 6 (36 samples), for the design of the calibration matrix, 22 samples were selected following a geometrical design as shown in figure 3; the remaining 14 samples were the individual spectra obtained in order to assure the fulfillment of Beer's law. The composition of the used mixtures to establish the calibration matrix is shown in Table 1.
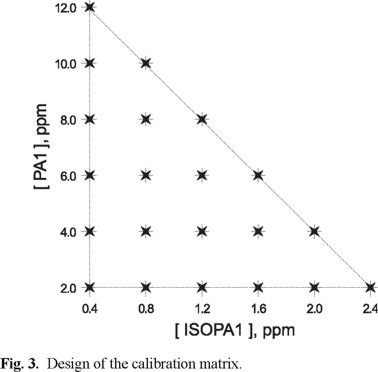
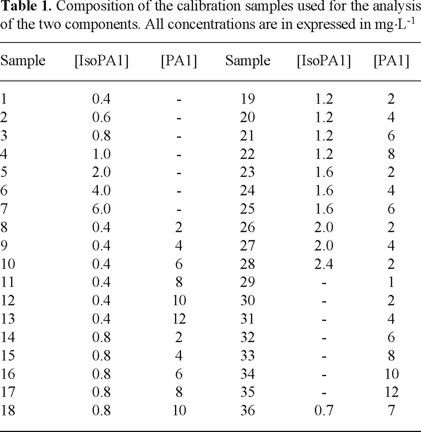
Once the experimental conditions were selected, the corresponding samples were prepared and the multicomponent algorithm PLS-1 was applied. This algorithm allowed the analysis of the compounds in the system in an independent way, starting off from the more relevant spectral information. Although the lineal relationship between the analytical property of interest (absorbance) and the concentration of each analyte in the multicomponent system is considered a priori, it is possible to evaluate by means of PLS-1, the variables affecting the prediction, such as deviation from linearity that could arise from interferences, noise, etc. On the other hand, PLS-1 allowed to establish the number of principal components necessary to construct the calibration model for each component. In this case, for the application of the algorithm in the simultaneous quantification of IsoPA1 and PA1, the computational program GRAMS/32tmv.5 from Galactic Co was used.
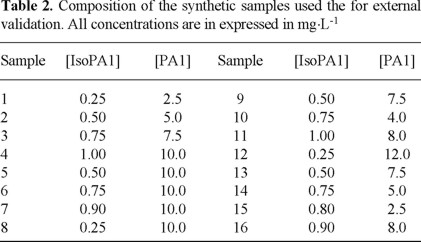
The data obtained from the calibration spectra in the whole range (1140 pair of data for each spectrum) were manipulated by means of the PLS-1 algorithm. The adequate selection of the number of factors used for the construction of the calibration matrix, is of critical importance for the correct application of this algorithm. The more commonly used method to make this selection is crossed validation: in each experiment one sample is left outside and the value PRESS (Prediction Residual Error Sum of Squares) is calculated for each resulting calibration model
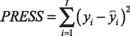
Where
yi is the known concentration of the analyte and
y is the estimated concentration of the analyte in each calibration sample.
In order to select the optimal number of factors, the minimal PRESS criterion or the statistical test F can be applied. In this last case an F value of 0.75 is the criterion commonly used [14]. It does not exist strict rules for using one or other of these criteria; we used both in this work.
On the other hand, in order to perform the internal and the external validation of the calibration model constructed, several statistical parameters were calculated. To perform the external validation, 16 synthetic mixtures of random composition were prepared. All samples were within the working concentration range proposed (Table 2).
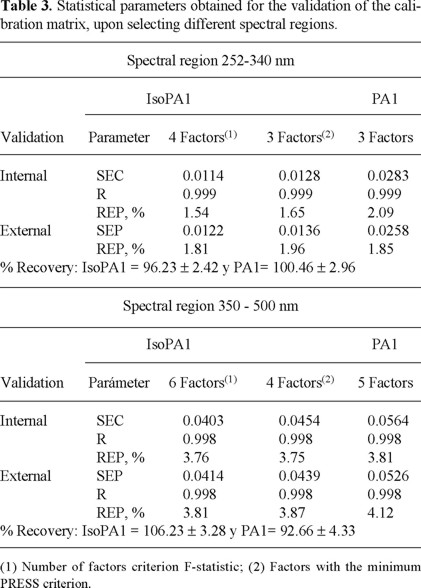
After the analysis of the results obtained from SEC and R2 values, using the full range spectra and taking into consideration both the minimum PRESS and the statistical test F, optimal factor numbers of 5 and 9 were found for PA1 and for IsoPA1 respectively. Nevertheless the prediction results were not totally satisfactory as % recovery for both compounds were 88 ± 21 %. For this reason, we decided to investigate the effect of the selection of the spectral region; besides the correction of the baseline, property that enhances the signal-noise relationship, was also evaluated.
The correct selection of the spectral regions, allowed the use of those wavelengths that appear more relevant for the calculations; the diminution of the number of wavelengths used must be avoided, because the inherent advantages of a multi-variate calibration method, namely the use of a great number of analytical signals to estimate the desired analytes, could be lost. In this case, the selection was made starting from the correlation spectra, which were calculated previously to the spectral decomposition; correlation spectra establish the lineal relationship between the analytical signal at each wavelength and the concentration of one of the components in the absorption spectra of the calibration samples.
The spectra of the substances under investigation are highly overlapped; therefore it is not surprising that only few signals gave high correlation coefficients. Following this criteria [20] two regions which showed the best correlation coefficients ranging between 350-500 nm (corresponding to 428 experimental points) and 252-340 nm (corresponding to 251 points), were selected. The obtained results are shown in Table 3; from these data it can be observed that, with the appropriate selection of the spectral regions, the recovery of both components is substantially improved. Therefore, upon the application of PLS-1, it is possible to undertake the simultaneous quantification of IsoPA1 and PA1 with satisfactory prediction ability, even in those mixtures where the concentration relationship is 25:1.
Real samples analysis
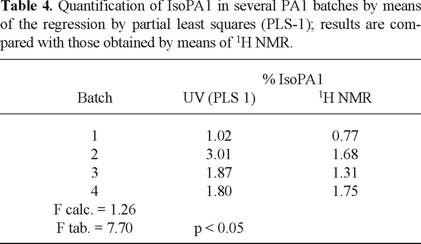
Finally, with the purpose of verifying the validity of the method here proposed, and after having established the optimal calibration model, the simultaneous determination of PA1 and isoPA1 in batches with different concentrations of IsoPA1 as determined by 1H NMR [8], was made. The obtained results are summarized in Table 4. Although the application of the experimental design here presented allowed the simultaneous quantification of PA1 and IsoPA1 when they are in a 25:1 relationship, in the real samples where a higher relationship is found, it was possible to quantify only the IsoPA1. Under the same conditions, the quantification of PA1 showed a remarkable subvaluation.
Conclusions
The multivariate calibration method, using partial least square regression, is a well known mathematical tool that can be applied in analytical chemistry when the analytical signals are highly overlapped and the relationship of the analytes concentrations is very large; in the current work, it was possible to calculate, with satisfactory results, the content of a contaminant present in concentrations lower than 1%.
Acknowledgments
Authors are acknowledged to CONACYT (México) for financial support in the form of a fellowship for M.L.S. and grants to N.W.T. and P.L.L.A.
References
1. Dreyer, D.; Arai, I.; Bachman, C.; Anderson, W.; Smith, R.; Daves, D. J. Am. Chem. Soc. 1975, 97, 4894-4990. [ Links ]
2. Piñeyro, A.; Martínez de Villarreal, L.; González Alanis, R. Toxicol. 1994, 92, 217-227. [ Links ]
3. Waksman-de-Torres, N.; Piñeyro-López, A. Chemistry, Structure and Biological Activity of Anthracenones of the Karwinskia genus. Studies in Natural Products Chemistry, Elsevier, editor Atta Ur Rahman, 2000, 22, 555-606. [ Links ]
4. Waksman, N.; Ramírez-Durón, R. Rev. Latinoamer. Quím. 1992, 23, 25-27 [ Links ]
5. Rivas, V.; Waksman, N. Nat. Prod. Lett. 2001, 15, 243-251. [ Links ]
6. A. Osorio-Pérez, A.; Salazar-Cavazos M.; Piñeyro-López, A.; Waksman de Torres, N. J. Chromatogr. 2003, 783, 85-92. [ Links ]
7. Salazar, M. L.;. López de Alba, P. L.; Piñeyro-López, A.; Waksman-de-Torres, N. J. Liq. Chromat. Rel. Tech. 2002, 25, 1747-1760. [ Links ]
8. Fernández-Ramírez, A.; Salazar-Cavazos, M.; Rivas-Galindo, V.; Ceniceros-Almaguer, L.; Waksman de Torres, N. Anal. Lett. 2004, 37, 2433-2444. [ Links ]
9. Grant, A.; Bhattacharyya, P. J. J. Chromatogr. 1985, 347, 219-235. [ Links ]
10. Fabre, H.; Le Bris, A.; Blanchin, M. D. J. Chromatogr. 1995, 697, 81-88. [ Links ]
11. Xueguang, S.; Wensheng C.; Peiyan S.; Maosen Z.; Guiwen Z. Anal. Chem. 1997, 69, 1722-1725. [ Links ]
12. Polster, J.; Sauerwald N.; Feucht W.; Treutter D. J. Chromatogr. 1998, 800, 121-137. [ Links ]
13. Savitzky, A.; Golay, J. E. Anal. Chem. 1964, 36, 1627-1639. [ Links ]
14. López-de-Alba.; P. L.; López-Martínez, L.; Hernández, A. Rev. Soc. Quím. Méx. 1996, 41, 34-44. [ Links ]
15. Guiberteau, A.; Galeano, T.; Espinosa, A.; López-de-Alba, P. L.; Salinas F. Anal. Chim. Acta. 1995, 302, 9-12. [ Links ]
16. Amador-Hernández, J.; Cladera, A.; Estela, J. M.; López-de-Alba, P. L.; Cerda, V. Analyst 1998, 123, 2215-2217. [ Links ]
17. Amador-Hernández, J.; López-Martínez, L.; Cladera, A.; Estela, J. M.; López-de-Alba, P. L.; Cerda, V. Bol. Soc. Chil. Quím. 1999, 44, 299-303. [ Links ]
18. López-de-Alba, P. L.; López-Martínez, L., González-Leal, M., Estrada-Hernández, Y. Quím. Anal. 1999, 18, 291. [ Links ]
19. Martens, H.; Naes, T. Multivariate Calibration, Wiley & Sons, New York, 1991. [ Links ]
20. Dubey, S. C.; Nadkarni, M. N. Talanta 1977, 24, 266-268. [ Links ]

