










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.48 no.4 Ciudad de México oct./dic. 2004
Investigación
Formal Synthesis of (±)-γ-Lycorane
Yazmín M. Osornio and Luis D. Miranda*
Instituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Coyoacán 04510, México, D.F. Tel.: 56-22-4440; fax: 56-16-2217. E-mail: lmiranda@servidor.unam.mx
Recibido el 17 de septiembre del 2004.
Aceptado el 17 de noviembre del 2004.
Abstract
A formal synthesis of (±)-γ-lycorane 4 based on the generation of the tetracyclic ketone 5 is disclosed. The general strategy of the synthesis of 5 rests on the rapid construction of the highly functionalized intermediates 6 or 20 by a xanthate-based radical addition-cyclization-oxidation cascade process, followed by either, a Friedel-Crafts type cyclization of 17 or a Dieckmann cyclization-decarboalkoxylation sequence on 21.
Key words: γ-lycorane, radical cyclization, alkaloids, xanthates.
Resumen
Se describe la síntesis formal del (±)-γ-licorano 4 en base a la formación de la cetona tetracíclica 5. La estrategia general de síntesis del compuesto 5 involucra la construcción de intermediarios altamente funcionalizados del tipo 6 o 20, utilizando un proceso en cascada vía radicales libres de adición/ciclación, seguido por una ciclación Friedel-Crafts de 17 y/o una reacción de ciclación/descarboxilación de Dieckmann de 21.
Palabras Clave: γ-licorano, radicales libres, alcaloides, xantatos.
This paper is dedicated with affection to the memory of Dr. Raymundo Cruz Almanza
Introduction
Lycorine-type alkaloids are characterized by a pyrrolo[d,e]phenanthridine skeleton 1 (galanthane ring system), and represent an important subclass within the large Amaryllidaceae alkaloid family [1]. Lycorine 2, and its reduced congeners α- an γ-lycorane 3 and 4, are representative of this large class of alkaloids. Several members of this alkaloid family possess potent biological activities such as antiviral, antineoplastic, insect antifeedant activity, plant growth inhibition, and disruption of protein synthesis [1,2]. Consequently, considerable effort has recently been devoted to the development of efficient synthetic approaches to these alkaloids and derivatives thereof [3]. Even though γ-lycorane 4 displays no apparent useful biological activity, it has become a popular target to showcase potential new strategies for the synthesis of lycorine-type alkaloids [3].
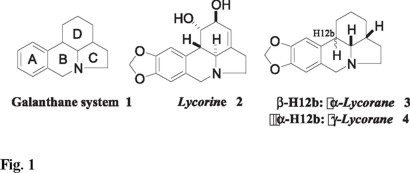
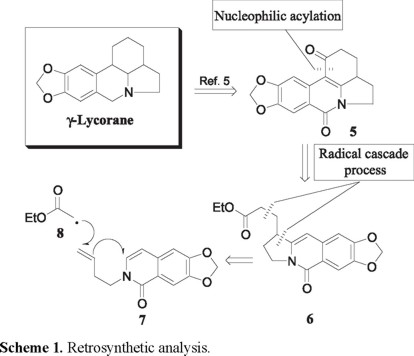
In connection with our ongoing interest in radical annulation reactions onto heterocyclic systems, we recently undertook a study of such processes in the isoquinolone area [4]. One consequence of this investigation was the demonstration that the radical cyclization of N-(3-iodoalkyl)isoquinolones constitutes a useful method for the rapid construction of the benzoindolizidine system, i.e., the A-B-C portion of the galanthane system 1 [4]. In this paper we describe a formal synthesis of (±)-γ-lycorane 4 wherein a radical cyclization to an isoquinolone nucleus is a central feature. The general strategy of the synthesis was to construct the advanced intermediate 5, wich transformation into 4 has already been described (Scheme 1) [5]. As illustrated by the retrosynthetic analysis (Scheme 1); the generation of tetracycle 5 was to be effected by a radical addition-cascade process on the N-butenylisoquinolone derivative 7, using the xanthate-based radical chemistry developed by Zard [6], followed by an intramolecular acylation of tricyclic 6 so produced, at the very nucleophilic C-4 carbon atom of the isoquinolone system.
Results and Discussion
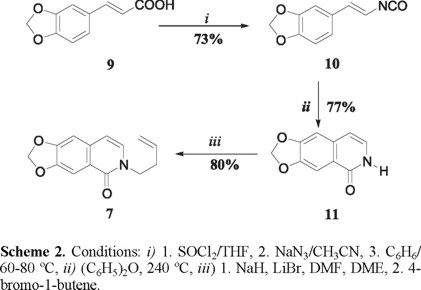
The synthesis of tricycle 6 started with the commercially available 3,4-(methylenedioxy)cinnamic acid 9, which was transformed into the isocyanate 10 by a standard Curtius procedure (Scheme 2). Thermal sigmatropic rearrangement of the isocyanate provided the isoquinolone 11 [7], which upon N-alkylation with 4-bromo-1-butene, by the Curran's method [8] generated the isoquinolone 7, the substrate for the desired sequential radical process.
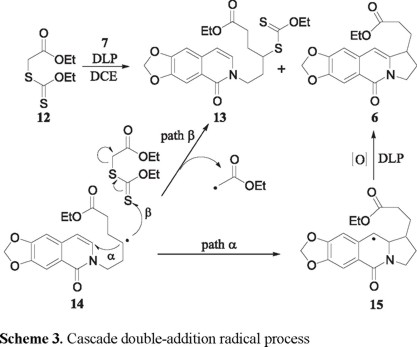
Heating a 1,2-dichloroethane (DCE) solution of 7 with equimolar amounts of the xanthate 12 and dilauroyl peroxide (DLP) at reflux temperature, produced a mixture of the desired tricyclic ester 6 (22 %, Scheme 3, path α), a new xanthate 13 (38 %), and starting material (15%). Compound 13 is obviously formed by a xanthate transfer mechanism [9] from 14 (path β), and can itself serve as a source of 6 if more DLP is added. Indeed, when the above reaction was carried out with 1.5 equivalents of DLP, the yield of 6 was raised to 55 %, while that of the xanthate 13 was reduced to 15 %. As expected, using two equivalents of the xanthate 12 in the presence of equimolar amounts of 6 and DLP markedly favored the formation of the xanthate transfer product 13 (45 %) over that of the ester 6 (17 %). It should be noted that in the conversion of 7 into 6, DLP serves as the source of the radicals 14 and 15 and also as an oxidant for the transformation of 15 into 6 [10].
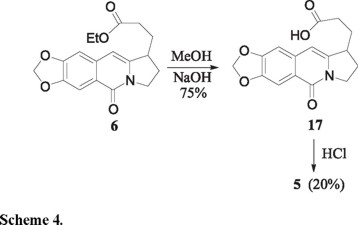
To complete the final phase in the intended construction of the galanthane skeleton 5, the ester 6 was saponified to the carboxylic acid 17 which cyclization was studied in detail (Scheme 4). Most mild Friedel-Crafts type conditions left the acid unchanged, and more vigorous ones destroyed it. Cyclization to 5 was successful only with 5M HCl, albeit in very modest yield (15 %).
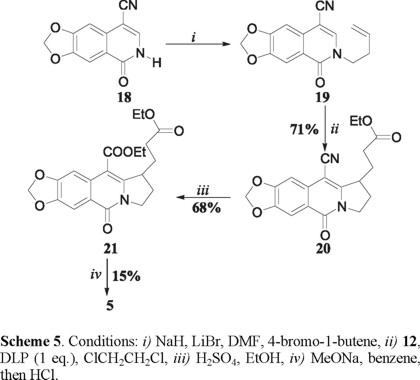
A Dieckmann cyclization-decarboalkoxylation strategy was also investigated in an attempt to devise a more efficient route to the tetracyclic ketone 5. N-butenylation of the known [11] 4-cyanoisoquinolone 18 provided 19 (Scheme 5) in good yield. Portionwise addition of DLP (1 equiv.) to an equimolar mixture of 19 and the xanthate 12 in boiling 1,2-dichloroethane solution produced the tricyclic cyanoester 20 in 71 % yield. None of the xanthate transfer product corresponding to 13 was observed. This no doubt stems from a diminished SOMO-LUMO energy gap [4a] in the acceptor, produced by the strong inductive effect of the nitrile moiety. The nitrile 20 was then transformed into the diester 21 by sulfuric acid catalyzed ethanolysis. Preliminary attempts to effect the required Dieckmann cyclization-decarboalkoxylation sequence on 21 were not promising so that the tetracycle 5 was once again produced in only 15 % yield. Notwithstanding, the low yield of 5 it can be readily converted into (±)-γ-lycorane [5]. The work described herein constitutes a formal synthesis of this alkaloid. Experiments to improve the efficiency for the transformation of 6 and 21 into 5 are in progress.
Conclusion
A formal synthesis of (±)-γ-lycorane 4 is described based on a four step secuence for the tetracyclic ketonic precursor 5. A central feature in the synthesis of 5 is the use of a cascade radical addition-cyclization-oxidation process to assemble highly functionalized intermediate progenitors of this tetracycle in one step from simple and readily available starting materials. It should be possible to apply the strategy described herein to the synthesis of a wide range of polycyclic alkaloids related to 4.
Experimental Section
The starting materials were purchased from Aldrich Chemical Co. and were used without further purification. Solvents were distilled before using them, ether and tetrahydrofuran were dried over sodium using benzophenone as indicator. Silica gel (230-400 mesh) was purchased from Merck. Silica plates of 0.20 mm thickness were used for thin layer chromatography. Melting points were determined with a Fisher-Johns melting point apparatus and they are uncorrected. 1H and 13C NMR spectra were recorded on either a Varian Gemini 200 MHz or Bruker 300 MHz spectrometer, the chemical shifts (δ) are given in ppm relative to TMS as internal standard (0.00). For analytical purposes the mass spectra were recorded on a JEOL JMS-5X 10217 in the EI mode, 70 eV, 200 °C via direct inlet probe. IR spectra were recorded on a Nicolet Magna 55-X FT instrument.
trans-β-(3,4-Methylenedioxy)styril isocyanate (10). A magnetically stirred solution of 3,4-(methylenedioxy)cinnamic acid 9 (1.8 g, 9.4 mmol) in THF (15 mL) maintained under a nitrogen atmosphere at 0 °C was added dropwise thionyl chloride (3.4 g, 2.1 mL, 28.1 mmol). After addition was complete the resulting mixture was heated at reflux for 3 h and then concentrated under reduced pressure. The residue was dissolved in anhydrous toluene (15 mL) and sodium azide (1.21 g, 18 mmol) was added at 0 °C. Stirring was continued for 0.5 h at 0 °C, then the reaction mixture was allowed to stir vigorously for a further 2 h at 60 °C. After this time the toluene was removed in vacuo. Distillation of the resulting pale yellow residue gave 10 (1.10 g, 73%) as a white solid, bp 130 °C (1.0 mmHg), mp 75-78 °C [7]; IR (KBr) νmax 2910, 2260, 1644 cm-1; RMN 1H (CDCl3, 300 MHz) δ 5.95 (2H, s), 6.33 (1H, d, J = 13.8 Hz), 6.43 (1H, d, J = 13.8 Hz), 6.70-6.80 (3H, m); MS (EI, 70 eV) m/z (%): 189 M+(100).
6,7-Methylenedioxy-2H-isoquinolin-1-one (11). A solution of trans-β-styril isocyanate 10 (1.4 g, 6.5 mmol) in diphenyl ether (20 mL) was heated at 240 ºC for 4 h. The isocarbostyril 11 was isolated by precipitation on cooling and by dilution with hexane. The solid (0.85 g, 77%), obtained, after trituration with hexane, was recrystallized from ethanol as needles, mp 283-284 ºC [7]; IR (KBr) νmax 2905, 2840, 1646, 1622, 1581 cm-1; RMN 1H (CDCl3, 300 MHz) δ 6.09 (2H, s), 6.37 (1H, d, J = 7.1 Hz), 6.97 (1H, d, J = 7.1 Hz), 7.0 (1H, s), 7.48 (1H, s), 11.09 (1H, br s); RMN 13C (CDCl3, 75 MHz) δ 101.2, 103.4, 104.6, 126.7, 136.9, 146.9, 151.2, 161.3; MS (EI, 70 eV) m/z (%): 189 M+(100).
2-(But-3-enyl)-6,7-methylenedioxy-2H-isoquinolin-1-one (7). To a suspension of NaH (0.11g, 4.4 mmol, 60% in mineral oil) in anhydrous DMF (1 mL) and DME (4 mL) was added a solution of the N-unsubstituted isoquinolone 11 (0.7 g, 3.7 mmol) in DME (1 mL) at 0 °C. After 10 min, the reaction mixture was treated with LiBr (0.64 g, 7.4 mmol) and stirred for 15 min, then 4-bromobutene (0.6 g, 0.45 mL, 4.5 mmol) in DME (2 mL) was added dropwise. The resulting mixture was stirred at room temperature for 4 h and then quenched with ice water. The mixture was extracted with ethyl acetate (3 × 15 mL), the combined organic layer was washed successively with water and saturated sodium chloride solution and dried over sodium sulfate. Removal of the solvent under reduced pressure followed by flash silica gel chromatography (hexane-ethyl acetate 7:3) gave 0.72 g (80%) of 7 as a white solid: mp 95-96 ºC; IR (KBr) νmax 2932, 2862, 1650, 1604, 1580 cm-1; RMN 1H (CDCl3, 300 MHz,) δ 2.49-2.57 (2H, m), 4.04 (2H, t, J = 7.2 Hz), 5.04-5.12 (2H, m), 5.72-5.87 (1H, m), 6.06 (2H, s), 6.35 (1H, d, J = 7.4 Hz), 6.84 (1H, s), 6.95 (1H, d, J = 7.4 Hz), 7.78 (1H, s); RMN 13C (CDCl3, 75 MHz,) δ 33.5, 48.9, 101.6, 103.6, 105.6, 105.8, 117.6, 121.8, 130.6, 134.2, 134.4, 147.8, 151.6, 161.2; MS (EI, 70 eV) m/z (%): 243 M+(50), 189 [M-54]+(100); HRMS (FAB+) calcd for C14H14O3N: 244.0974, found: 244.0975.
4-Ethoxythiocarbonylsulfanyl-6-(1-oxo-6,7-methylenedioxy-1H-isoquinolin-2-yl)-hexanoic acid ethyl ester (13) and 3-(7,8-Methylenedioxy-5-oxo-1,2,3,5-tetrahydropyrrolo[1,2-b]isoquinolin-1-yl)-propionic acid ethyl ester (6). To a degassed solution of 7 (0.5 g, 2.1 mmol) and 12 (0.64 g, 3.1 mmol) in refluxing dichloroethane (7 mL/mmol) was added lauroyl peroxide (1.22 g, 3.1 mmol) portionwise (0.5 mmol/1.5 h). The reaction was carried out under an atmosphere of N2 during 6.0 h. Then, the mixture was allowed to warm to room temperature and evaporated to dryness. The crude residue was purified by column chromatography on silica gel using hexane-ethyl acetate (7:3) to give 0.15 g (16%) of 13 as a yellow oil and 0.35 g (53%) of 6 as a white solid mp 129-130 ºC (dec).
Compound 13. IR (CHCl3) νmax 2919, 2850, 1727, 1649, 1625, 1599 cm-1; RMN 1H (CDCl3, 300 MHz,) δ 1.25 (3H, t, J = 7.2 Hz), 1.38 (3H, t, J = 7.2 Hz), 1.88-2.35 (4H, m), 2.45-2.58 (2H, m), 3.78-3.84 (1H, m), 4.11 (2H, q, J = 7.2 Hz), 4.08-4.18 (2H, m), 4.60 (2H, q, J = 7.2 Hz), 6.08 (2H, s), 6.34 (1H, d, J = 7.4 Hz), 6.86 (1H, s), 6.96 (1H, d, J = 7.4 Hz), 7.79 (1H, s); RMN 13C (CDCl3, 75 MHz,) δ 14.2, 28.1, 28.4, 31.7, 42.3, 46.6, 60.7, 100.4, 101.6, 103.5, 105.0, 120.2, 135.4, 145.1, 147.2, 151.8, 160.6, 172.8; MS (EI, 70 eV) m/z (%): 329 M+(45), 284 [M-54]+(100); HRMS (FAB+) calc for C21H26O6S2N: 452.1201, found: 452.1205.
Compound 6. IR (KBr) νmax 2944, 2914, 1731, 1649, 1627, 1475 cm-1; RMN 1H (CDCl3, 300 MHz,) δ 1.28 (3H, t, J = 7.2 Hz), 1.73-1.94 (2H, m), 2.14-2.50 (4H, m), 3.20-3.31 (1H, m), 3.93-4.34 (2H, m), 4.14 (q, 2H, J = 7.2 Hz), 6.06 (2H, s), 6.30 (1H, s), 6.83 (1H, s), 7.72 (1H, s); RMN 13C (CDCl3, 75 MHz,) δ 14.2, 28.1, 28.4, 31.7, 42.3, 46.6, 60.7, 100.4, 101.6, 103.5, 105.0, 120.2, 135.4, 145.1, 147.2, 151.8, 160.6, 172.8; MS (EI, 70 eV) m/z (%): 329 M+(45), 284 [M-54]+(100); HRMS (FAB+) calcd for C18H20O5N: 330.1341, found: 330.1340.
3-(7,8-Methylenedioxy-5-oxo-1,2,3,5-tetrahydropyrrolo[1,2-b]isoquinolin-1-yl)-propionic acid (17). A magnetically stirred solution of the ester 6 (0.3 g, 0.9 mmol) and NaOH (0.11 g, 2.7 mmol) in aqueous methanol (2 mL of a 4:3 v/v mixture) was heated at reflux for 16 h. The cooled mixture was then acidified with HCl (conc. aqueous solution) and extracted with CH2Cl2 (4 × 10 mL). The combined organic phases were dried, filtered and concentrated under reduced pressure to give the acid 17 (0.20 g, 75%) as a light-yellow oil. This material was used without further purification in the next step of the reaction sequence. IR (CHCl3) νmax 3226, 2926, 1713, 1644, 1584 cm-1; RMN 1H (CDCl3, 300 MHz,) δ 1.70-1.96 (2H, m), 2.14-2.50 (4H, m), 3.25-3.32 (1H, m), 4.04-4.15 (1H, m), 4.33 (1H, ddd, J = 3.2, 9.2 and 12.6 Hz), 6.08 (2H, s), 6.30 (1H, s), 6.83 (1H, s), 7.72 (1H, s); MS (EI, 70 eV) m/z (%): 301 M+(65), 284 [M-17]+(100); HRMS (FAB+) calcd for C16H16O5N: 302.1028, found: 302.1034.
3,3a,4,5-Tetrahydro-9,10-methylenedioxy-2H-pyrrolo[3,2,1-de]phenanthridine-1,7-dione (5). A magnetically stirred solution of the acid 17 (0.15 g, 0.5 mmol) in a 5 M HCl aqueous solution (4 mL) was mantained at 60 °C for 3 h, then made alkaline by the addition of Na2CO3 (saturated aqueous solution) and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried, filtered and concentrated under reduced pressure to give a light-yellow oil. This material was purified by column chromatography on silica gel using hexane-ethyl acetate (6:4) to give 28 mg (20%) of 5 as a white solid: mp 249-250 ºC. Spectra data were identical with those reported before [5]. IR (KBr) νmax 2970, 2911, 1709, 1645, 1620, 1592 cm-1; MS (EI, 70 eV) m/z (%): 283 M+(100); HRMS (FAB+) calcd for C16H14O4N: 284.0923, found: 284.0927.
2-(But-3-enyl)-4-cyano-6,7-methylenedioxy-2H-isoquinolin-1-one (19). To a suspension of NaH (0.08 g, 3.6 mmol, 60% in mineral oil) in anhydrous DMF (1 mL) and DME (4 mL) was added a solution of the nitrile 18 [11] (0.8 g, 3.7 mmol) in DME (1 mL) at 0 °C. After 10 min, the reaction mixture was treated with LiBr (0.64 g, 7.4 mmol) and stirred for 15 min, then a solution of 4-bromobutene (0.6 g, 0.45 mL, 4.5 mmol) in DME (2 mL) was added dropwise. The resulting mixture was stirred at room temperature for 4 h and then quenched with ice water. The mixture was extracted with ethyl acetate (3 × 15 mL), the combined organic layer was washed successively with water and saturated sodium chloride solution and dried over sodium sulfate. Removal of the solvent under reduced pressure followed by flash silica gel chromatography (hexane-ethyl acetate 7:3) gave 0.70 g (70%) of 19 as a white solid: mp 142-144 ºC; IR (KBr) νmax 2914, 2216, 1654, 1619, 1583 cm-1; RMN 1H (CDCl3, 300 MHz,) δ 2.51-2.58 (2H, m), 4.07 (2H, t, J = 7.0 Hz), 5.04-5.12 (2H, m), 5.72-5.85 (1H, m), 6.15 (2H, s), 7.15 (1H, s), 7.57 (1H, s), 7.74 (1H, s); RMN 13C (CDCl3, 75 MHz,) δ 33.2, 49.7, 90.4, 102.2, 102.4, 106.1, 115.9, 118.7, 120.9, 130.8, 133.3, 139.2, 149.0, 153.0, 160.2; MS (EI, 70 eV) m/z (%): 268 M+(40), 214 [M-54]+(100); HRMS (FAB+) calcd for C15H13O3N: 269.0926, found: 269.0920.
Ethyl-3-(10-Cyano-7,8-methylenedioxy-5-oxo-1,2,3,5-tetrahydropyrrolo[1,2-b]isoquinolin-1-yl) propanoate (20). To a degassed solution of 19 (0.4 g, 1.5 mmol) and 12 (0.47 g, 2.2 mmol) in refluxing dichloroethane (7 mL/mmol) was added lauroyl peroxide (0.59 g, 1.5 mmol) portionwise (0.3 mmol/1.5 h). The reaction was carried out under an atmosphere of N2 during 5.0 h. Then, the mixture was allowed to cool to room temperature and evaporated to dryness. The crude residue was purified by column chromatography on silica gel using hexane-ethyl acetate (6:4) to give 0.38 g (71%) of 20 as a white solid mp 148-149 ºC; IR (KBr) νmax 2975, 2914, 2211, 1728, 1648, 1622, 1582 cm-1; RMN 1H (CDCl3, 300 MHz,) δ 1.26 (3H, t, J = 7.2 Hz), 1.98-2.12 (2H, m), 2.26-2.45 (2H, m), 2.49 (2H, t, J = 7.0 Hz), 3.56-3.72 (1H, m), 4.04-4.19 (1H, m), 4.14 (q, 2H, J = 7.2 Hz), 4.32 (1H, ddd, J = 3.5, 9.0 and 12.3 Hz), 6.13 (2H, s), 7.17 (1H, s), 7.69 (1H, s); RMN 13C (CDCl3, 75 MHz,) δ 14.0, 26.5, 27.8, 31.7, 43.7, 47.5, 60.7, 85.5, 101.8, 102.3, 105.2, 115.5, 119.4, 131.9, 148.3, 153.0, 155.2, 159.4, 172.1; MS (EI, 70 eV) m/z (%): 354 M+(40), 266 [M-88]+(100); HRMS (FAB+) calcd for C19H19O5N2: 355.1294, found: 355.1292.
Ethyl-3-(10-Carbomethoxy-7,8-methylenedioxy-5-oxo-1,2,3,5-tetrahydropyrrolo[1,2- b ]isoquinolin-1-yl)propanoate (21). A solution of 20 (0.3 g, 0.85 mmol) in 8 mL of EtOH and 3 mL of concentrated sulphuric acid was refluxed for 8 h. The product was extracted into ethyl acetate and the extract was washed with water and brine, dried over Na2SO4 and concentrated at reduced pressure to give 21 (0.23 g, 68%) as a white solid: mp 192-193 ºC; IR (KBr) νmax 2975, 2914, 2211, 1728, 1648, 1622, 1582 cm-1; RMN 1H (CDCl3, 300 MHz,) δ 1.42 (3H, t, J = 7.2 Hz), 1.47 (3H, t, J = 7.2 Hz), 2.03-2.09 (2H, m), 2.31-2.45 (4H, m), 3.89-4.23 (2H, m), 4.13 (q, 2H, J = 7.2 Hz), 4.38-4.45 (2H, m), 6.08 (2H, s), 7.76 (1H, s), 8.25 (1H, s); MS (EI, 70 eV) m/z (%): 401 M+(90), 268 [M-133]+(100); HRMS (FAB+) calcd for C21H24O7N: 402.1553, found: 402.1549.
Acknowledgments
We thank DGAPA-UNAM (PAPIIT-IN228103) for generous financial support. Also we thank Rocío Patiño, Elizabeth Huerta, Angeles Peña, Javier Pérez, Luis Velasco and Nieves Zavala, for their technical support.
References
1. For general reviews on lycorine-type alkaloids see: (a) Hoshino, O. In The Alkaloids: Cordell, G. A. Ed.; Academic Press: New York, 1998; Vol. 51, pp 323-424. [ Links ] (b) Martin, S. F. In The Alkaloids: Brossi, A. Ed.; Academic Press: New York, 1987; Vol. 30, pp 251-376. [ Links ]
2. (a) Leven, M.; Van den Berghe, D. A.; Vlietnck, A. J. Planta Med. 1983, 49, 109. [ Links ] (b) Pettit, G. R.; Gaddamidi, V.; Goswami, A.; Cragg, G. M. J. Nat. Prod. 1984, 47, 796-801. [ Links ]
3. (a) Sugiyama, N.; Narimiya, M.; Iida, H.; Kikuchi, T. J. Heterocycl. Chem. 1988, 25, 1455-1457. [ Links ] (b) Bäckvall, J-E.; Andersson, P. G.; Stone, G. B.; Gogoll, A. J. Org. Chem. 1991, 56, 2988-2993. [ Links ] (c) Pearson, W. H.; Schkeryantz, J. M. J. Org. Chem. 1992, 57, 6783-6789. [ Links ] (d) Grotjahn, D. B.; Vollhardt, K. P. C. Synthesis, 1993, 579-605. [ Links ] (e) Banwell, M. G.; Wu, A. W. J. Chem. Soc.,Perkin Trans.1 1994, 2671-2672. [ Links ] (f) Yoshizaki, H.; Satoh, H.; Sato, Y.; Nukui, S.; Shibasaki, M.; Mori, M. J. Org. Chem. 1995, 60, 2016-2021. [ Links ] (g) Angle, S. R.; Boyce, J. P. Tetrahedron Lett. 1995, 36, 6185-6188. [ Links ] (h) Cassayre, J.; Zard, S. Z. Synlett. 1999, 501-503. [ Links ] (i) Hoang-Cong, X.; Quiclet-Sire, B.; Zard, S. Z. Tetrahedron Lett. 1999. 40, 2125-2126. [ Links ] (j) Cossy, J.; Tresnard, L.; Pardo, D. G. Tetrahedron Lett. 1999, 40, 1125-1128. [ Links ] (k) Banwell, M. G.; Harvey, J. E.; Hockless, D. C. R. J. Org. Chem. 2000, 65, 4241-4250. [ Links ] (l) Padwa, A.; Brodney, M. A.; Lynch, S. M. J. Org. Chem. 2001, 66, 1716-1724. [ Links ] (m) Yasuhara, T.; Osafune, E.; Nishimura, K.; Yamashita, M.; Yamada, K-I.; Muraoka, O.; Tomioka, K. Tetrahedron Lett. 2004, 45, 3043-3045. [ Links ]
4. (a) Osornio, Y. M.; Miranda, L. D.; Cruz-Almanza, R.; Muchowski, J. M. Tetrahedron Lett. 2004, 45, 2855-2858. [ Links ] (b) Menes-Arzate, M.; Martinez, R.; Cruz-Almanza, R.; Muchowski, J. M.; Osornio, Y. M.; Miranda, L. D. J. Org. Chem. 2004, 69, 4001-4004. [ Links ]
5. (a) Iida H.; Aoyagi, S.; Kibayashi, C. J. Chem. Soc., Perkin Trans 1. 1975, 2502-2506. [ Links ] (b) Iida, H.; Yuasa, Y.; Kibayashi, C. J. Org. Chem. 1979, 44, 1074-1080. [ Links ]
6. For reviews of this reaction see: (a) Zard, S. Z. in "Radicals in Organic Synthesis", Ed. P. Renaud and M. Sibi, Wiley VCH, Weinhem, 2001, p. 90-108. [ Links ] (b) Quiclet-Sire, B.; Zard, S. Z. Phosphorus, Sulfur, Silicon., 1999, 153-154, 137. [ Links ] (c) Zard, S. Z. Angew. Chem. Int. Ed. Engl. 1997, 36, 672-685, [ Links ] and references cited therein.
7. (a) Briet, N.; Brookes, M. H.; Davenport, R. J.; Galvin, F. C. A.; Gilbert, P. J.; Mack, S. R.; Sabin, V. Tetrahedron 2002, 58, 5761-5766. [ Links ] (b) Mikol, G. J.; Boyer, J. H. J. Org. Chem. 1972, 37, 724-726. [ Links ]
8. (a) Curran, D. P.; Yu, H.; Liu, H. Tetrahedron 1994, 50, 7343-7366. [ Links ] (b) Curran, D. P.; Liu, H. J. Chem. Soc., Perkin Trans. 1. 1994, 1377-1393. [ Links ]
9. (a) Kaoudi, T.; Miranda, L. D.; Zard, S. Z. Org. Lett. 2001, 3; 3125-3127. [ Links ] (b) Bertrand, F.; Pevere, V. Quiclet-Sire, B. Zard, S. Z. Org. Lett. 2001, 3, 1069-1071. [ Links ] (c) Gagosz, F.; Zard, S. Z. Org. Lett. 2002, 4, 4345-4348. [ Links ] (d) Gagosz, F.; Zard, S. Z. Org. Lett. 2003, 5, 2655-2657. [ Links ]
10. (a) Axon, J.; Boiteau, L.; Boivin, J.; Forbes, J. E. and Zard, S. Z. Tetrahedron Lett. 1994, 35, 1719-1722. [ Links ] (b) Liard, A.; Quiclet-Sire, B.; Saicic, R. N.; Zard, S. Z. Tetrahedron Lett. 1997, 38, 1759-1762. [ Links ] (c) Cholleton, N.; Zard, S. Z. Tetrahedron Lett. 1998, 39, 7295-7298. [ Links ] (d) Ly, T. M.; Quiclet-Sire, B.; Sortais, B.; Zard, S. Z. Tetrahedron Lett. 1999, 40, 2533-2536. [ Links ] (e) Kaoudi, T.; Quiclet-Sire, B.; Seguin, S.; Zard, S. Z. Angew. Chem. Int. Ed. 2000, 39, 731-733. [ Links ] (f) Osornio, Y. M.; Cruz-Almanza, R.; Jiménez-Montaño, V.; Miranda, L. D. Chem. Commun. 2003, 2316-2317. [ Links ] See also ref. [4b].
11. Passannanti, S.; Paternostro, M. P.; Piozzi, F.; Savona, G. J. Heterocyclic. Chem. 1977, 14, 103-106. [ Links ]

