










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.48 no.4 Ciudad de México oct./dic. 2004
Investigación
Estudio Sintético de Furanoeremofilanos. Síntesis de la 13-Nor-9-oxoeuryopsina
José Agustín Guzmán,1* Esther García,1 Virgilio Mendoza,1 Damaristelma de Jesús1 y Luis Ángel Maldonado2*
1 Instituto de Investigaciones Químico-Biológicas, UMSNH, Edificio B-1, Ciudad Universitaria, 58030 Morelia, Mich.; Tel. (443)-326-5790. E-mail: guzbar@zeus.umich.mx
2 Instituto de Química de la Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, 04510 México, D.F.; Tel. 5622-4428. E-mail: lammg@servidor.unam.mx
Recibido el 10 de septiembre del 2004.
Aceptado el 26 de octubre del 2004.
Resumen
En este artículo describimos una estrategia diseñada para la síntesis del esqueleto básico de los furanoeremofilanos, los cuales constituyen la clase más abundante entre los furanosesquiterpenos. Las etapas clave son la doble alquilación regio- y estereoselectiva de una enona (difuncionalización vecinal en tándem) y la construcción del anillo central mediante una reacción de sustitución electrofílica aromática intramolecular. Para demostrar la factibilidad de nuestra propuesta, sintetizamos el norfuranoeremofilano 1 en 8 etapas, utilizando como materias primas 2-metil-2-ciclohexenona y 3-furaldehído.
Palabras clave: Síntesis de furanoeremofilanos, 13-nor-9-oxoeuryopsina, difuncionalización vecinal en tándem.
Abstract
In this paper we describe a strategy designed for the synthesis of the basic skeleton of furanoeremophilanes, the most abundant class of furanosesquiterpenes. The key steps in the synthetic scheme involve a regio- and stereoselective double alkylation of an enone (tandem vicinal difunctionalization) and construction of the central ring via an intramolecular electrophilic aromatic substitution. To demonstrate this approach, we synthesized the norfuranoeremophilane 1 in 8 steps, starting from 2-methyl-2-cyclohexenone and 3-furaldehyde.
Key words: Furanoeremophilane synthesis, 13-nor-9-oxoeuryopsin, tandem vicinal difunctionalization.
Dedicado a la memoria del doctor Raymundo Cruz Almanza
Introducción
Los furanoeremofilanos constituyen el grupo más numeroso de los sesquiterpenos que contienen un anillo furánico [1], encontrándose ampliamente distribuidos en plantas de varios géneros (Euryops, Ligularia, Psacalium, Senecio, entre otros) [2,3] pertenecientes a la familia Asteraceae. Las características estructurales comunes a todos los miembros de esta clase de productos naturales son: un sistema tricíclico de decalin[2,3-b]furano, dos metilos syn en los átomos de carbono C-4a y C-5 y un metilo unido a C-3 (Fig. 1). Adicionalmente pueden presentar diversos grados de oxidación en varias posiciones del sistema decalínico, el cual generalmente prefiere la fusión cis.
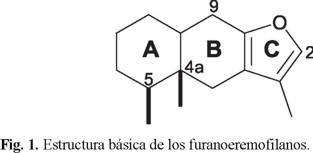
La interesante estructura de estos compuestos, así como las actividades biológicas mostradas por algunos de ellos, ha atraído la atención de muchos químicos sintéticos, de tal forma que se han descrito en la literatura algunas estrategias exitosas para su síntesis [4-9]. Por otro lado, desde el punto de vista estereoquímico, la dificultad más importante para la síntesis de la estructura básica de los furanoeremofilanos radica en el control de la configuración relativa syn de los metilos unidos a C-4a y C-5 del sistema tricíclico.
Para demostrar la factibilidad de una estrategia sintética que podría ser utilizada para la preparación de varios furanoeremofilanos de origen natural, en este trabajo presentamos la síntesis de (4aR*, 5S*)-4a,5-dimetil-4a,5,6,7-tetrahidro-4H-nafto[2,3-b]furan-9-ona (1), compuesto que posee la mayoría de las características estructurales de estos productos naturales. La etapa clave consistió en una alquilación-hidroxialquilación regio- y estereoselectiva en tándem sobre la 2-metil-2-ciclohexenona, mediante la cual se logró el establecimiento y control de la configuración relativa de dos centros estereogénicos.
Resultados y discusión
La preparación regioselectiva de cetonas α,β-disustituidas ha sido un objetivo sintético importante, debido a que dicho agrupamiento se encuentra frecuentemente en productos de origen natural. Uno de los procedimientos más convenientes para lograrlo consiste en la adición conjugada de un organocuprato a una cetona α,β-insaturada y atrapar in situ el enolato así formado con un electrófilo apropiado [10] (Esquema 1). El enorme potencial sintético de esta secuencia (difuncionalización vecinal en tándem) se ha hecho evidente en su aplicación como etapa importante, en la síntesis de productos naturales como prostaglandinas [11], sesquiterpenos [12] y esteroides [13], entre otros.
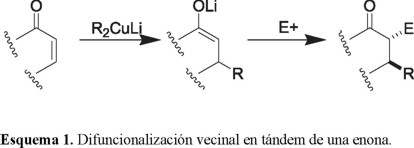
Basados en este proceso, diseñamos una estrategia para la síntesis del esqueleto básico de los furanoeremofilanos en la cual el enolato 2, generado por la adición conjugada de dimetilcuprato de litio a 2-metil-2-ciclohexenona, puede ser atrapado con el 4-metil-3-furaldehído produciendo el cetol 3 (Esquema 2). De esta manera, si como ocurre en procesos similares, los dos grupos que se adicionan consecutivamente a la enona quedan situados en lados opuestos [10], esto dará como resultado que los dos metilos presenten una orientación syn, tal y como se indica en 3. Finalmente, la formación del sistema tricíclico completaría la secuencia sintética.
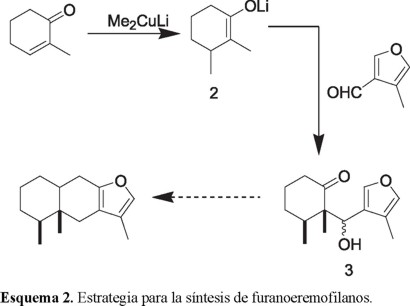
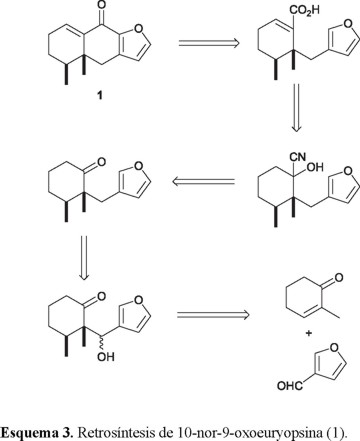
La 9-oxoeuryopsina es un furanoeremofilano típico aislado de varias especies de Euryops y Senecio [14, 15] y, a fin de someter a prueba la estrategia diseñada, decidimos intentar la síntesis del derivado 13-nor-9-oxoeuryopsina 1, cuyo análisis retrosintético se muestra en el Esquema 3. La razón de seleccionar como objetivo sintético un norfuranoeremofilano en lugar de un producto natural, residió en la disponibilidad comercial del aldehído furánico requerido como materia prima.
Después de optimizar algunas variables de reacción, logramos controlar y reproducir el curso de la primera y fundamental etapa de la síntesis (Esquema 4), consistente en la adición conjugada de dimetilcuprato de litio (2 eq. molar, -5 °C, 0.5 h) sobre la 2-metil-2-ciclohexenona [16] y atrapar el enolato con 3-furaldehído (1.5 eq. molar, -5 °C, 0.25 h). Esta secuencia produjo en buen rendimiento (86%) una mezcla estereoisomérica de los cetoles 4a y 4b en relación 2:1, fácilmente separables por métodos cromatográficos.
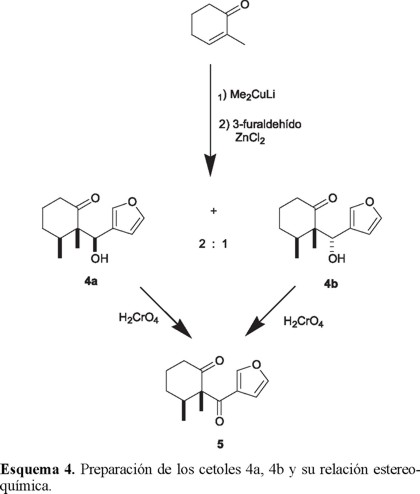
Las estructuras de estos cetoles fueron determinadas mediante las técnicas analíticas usuales y su relación epimérica fue inferida por la oxidación de cada diasterómero a la misma β-dicetona 5 (Esquema 4).
La configuración relativa syn de los dos grupos metilo fue establecida de manera inequívoca en un trabajo previo [17] a través del análisis de difracción de rayos-X del cetol estereomérico cristalino 4b, pf 87-88 °C.
La siguiente transformación consistió en la sustitución del grupo alcohol por hidrógeno de los cetoles 4a y 4b al mismo compuesto 6. Aunque en la literatura están descritos numerosos métodos para lograr este tipo de desoxigenaciones [18], la aplicación de algunos de estos procedimientos a nuestro caso particular no fue satisfactoria y sólo condujo a productos de procesos retroaldólicos o mezclas complejas. Finalmente, pudimos llevar a cabo esta desoxigenación utilizando el método de Barton-McCombie [19]. Así, el tratamiento de los cetoles 4a y 4b (individualmente o como mezcla) con un exceso (2.5 eq. molar) de tiocarbonildiimidazol (TCDI), seguido de la reducción del tiocarbamato producido (no caracterizado) con hidruro de tri(n-butil)estaño (n-Bu3SnH), en presencia de 2,2'-azobisisobutironitrilo (AIBN) como catalizador, produjo en rendimiento aceptable (55%) un aceite transparente y casi incoloro, el cual fue identificado como la cetona 6 (Esquema 5). La ausencia de la banda característica del grupo hidroxilo en el espectro IR y la presencia en el espectro de RMN 1H de un sistema AB centrado en δ 2.71 (J = 15 Hz), asignado al metileno vecino al anillo furánico, fueron determinantes en la caracterización de 6.
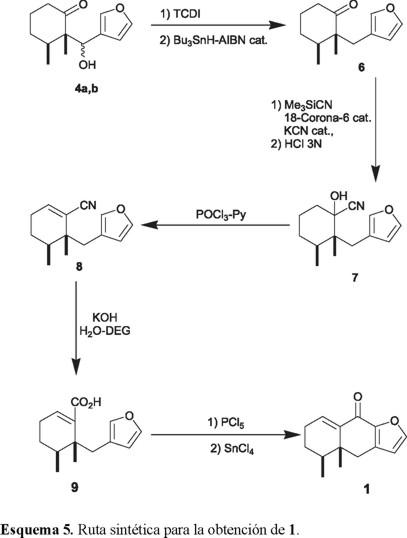
Para introducir el átomo de carbono necesario para la formación del sistema tricíclico, el compuesto 6 se trató de convertir en la cianohidrina 7 con excesos de KCN y AcOH, pero sin éxito. Aparentemente factores estéricos en 6 ocasionan un equilibrio desfavorable en esta reacción reversible. Posteriormente se encontró que un procedimiento indirecto, en el cual 6 se trató con un pequeño exceso (1.5 eq. molar) de cianuro de trimetilsililo (Me3SiCN) y cantidades catalíticas de KCN y éter 18-corona-6 en benceno a t.a. durante una hora [20], dio lugar a la correspondiente cianohidrina O-trimetilsililada, la cual fue hidrolizada en medio ácido (HCl 3N, 3 h) al α-hidroxinitrilo 7 requerido en 93% de rendimiento para los dos pasos. Debido a la presencia de un nuevo centro estereogénico, este último intermediario fue obtenido como una mezcla de dos diasterómeros.
La deshidratación de la cianohidrina 7 (POCl3/Py, 100 °C, 2 h), produjo el nitrilo α,β-insaturado 8 (70% de rendimiento) como un aceite incoloro. En su espectro IR se observó la banda característica del grupo nitrilo en 2214 cm-1 con una alta intensidad, debido a su conjugación con el doble enlace del ciclohexeno. La hidrólisis de 8 en medio básico (KOH en H2O-dietilenglicol, 72 h a reflujo) produjo el ácido carboxílico 9 y una fracción neutra, que aunque no se investigó, debe contener la amida intermediaria. La repetición del tratamiento básico a reflujo antes citado sobre esta fracción neutra produjo una cantidad adicional de 9 y otra fracción neutra la cual volvió a ser tratada de la misma forma. El rendimiento de 9 crudo obtenido después de estos tres tratamientos básicos fue de 72 % y se caracterizó espectroscópicamente como el éster metílico (CH2N2 en éter).
La última etapa de la síntesis requirió la conversión de 9 en su correspondiente cloruro de ácido (PCl5/benceno, 5 °C→t.a., 1.25 h), seguida de una acilación intramolecular de Friedel y Crafts usando cloruro de estaño (IV) como ácido de Lewis (SnCl4/benceno, 5 °C, 0.5 h). El producto crudo de esta secuencia de reacciones, obtenido como un aceite rojizo, se purificó por cromatografía en columna para dar cristales blancos, pf 150-151 °C (de éter-hexano). El análisis de sus datos espectroscópicos (IR, RMN 1H y 13C, EM), así como su análisis elemental, fueron concordantes con la estructura de la enona tricíclica 1.
Conclusiones
La mayoría de las estrategias utilizadas para la síntesis de furanoeremofilanos, se basan en la construcción inicial del sistema decalínico y la posterior introducción del anillo furánico (ruta AB→ABC) o bien en la preparación inicial de un sistema hidrobenzofuránico seguida de la construcción del anillo A (ruta BC→ABC). Los resultados obtenidos en este trabajo demuestran que es posible la síntesis de furanoeremofilanos, conectando inicialmente los anillos A y C del sistema tricíclico al mismo tiempo que se establece la estereoquímica relativa de los dos grupos metilo y posteriormente la construcción del anillo central por medio de una sustitución electrofílica aromática intramolecular (ruta AC→ABC).
En principio, con los ajustes necesarios en la sustitución del aldehído furánico de partida y en las reacciones necesarias para cada caso particular, la estrategia mostrada en la presente investigación debe ser de utilidad en la síntesis de un número importante de furanoeremofilanos de origen natural.
Parte experimental
Las reacciones involucrando reactivos sensibles al oxígeno y/o humedad fueron realizadas bajo atmósfera seca de argón o nitrógeno. Los disolventes fueron secados utilizando técnicas descritas en la literatura. Las purificaciones por cromatografía en columna fueron hechas por gravedad usando como adsorbente gel de sílice Merck de 70-230 mallas. Los puntos de fusión fueron determinados en un aparato Fisher-Johns y se dan sin corregir. La espectroscopía IR fue obtenida en un equipo Perkin Elmer Spectrum One. Los espectros de RMN 1H (200 MHz) y 13C (50 MHz) se determinaron en un equipo Varian Gemini 200, usando CDCl3 como disolvente. Los desplazamientos químicos (δ) se dan en ppm relativos al TMS usado como referencia interna. Los análisis elementales fueron realizados en el Departamento de Química de la Universidad Autónoma Metropolitana, Unidad Iztapalapa. Los espectros de masas se obtuvieron en un equipo Hewlett Packard 5989B.
Preparación de 4a y 4b
Se agregó gota a gota una solución de 2-metil-2-ciclohexenona (1.65 g; 15 mmol) en éter anhidro (15 mL) a una solución en agitación de dimetilcuprato de litio (30 mmol) en éter anhidro (200 mL) a -5 °C bajo una atmósfera de nitrógeno seco, formándose rápidamente un precipitado amarillo. Después de 0.5 h, se adicionó una solución etérea 1 M de cloruro de zinc (30 mL) y después una solución de 3-furaldehído (1.8 g; 18.75 mmol) en éter anhidro (15 mL). Se continuó la agitación por 0.25 h a -5 °C y entonces la mezcla se vertió en 350 mL de una solución en agitación de NH4Cl acuoso al 20%. Se separó la fase orgánica y la fase acuosa, color azul intenso, se extrajo con éter (2 × 50 mL); la fase etérea reunida se lavó consecutivamente con soluciones acuosas de NH4Cl al 10% (2 × 30 mL) y saturada de NaCl (2 × 30 mL). Después de secar (Na2SO4) y evaporar el disolvente orgánico, el producto crudo se separó por cromatografía en columna (hexano/AcOEt, 6:1), obteniéndose 1.94 g (58%) de 4a como un aceite incoloro y 0.93 g (28%) de 4b como un sólido blanco cristalino, pf 87-88 °C (prismas de éter-hexano). 4a: IR (película) νmáx 3479, 1694 cm-1; RMN 1H (CDCl3) δ 7.35 (2H, m), 6.32 (1H, m), 4.51 (1H, sa), 2.53-2.42 (1H, m), 2.33-2.25 (1H, m), 1.95-1.76 (2H, m), 1.62-1.5 (3H, m), 1.35 (3H, s), 0.89 (3H, d, J = 7 Hz); RMN 13C (CDCl3) δ 219.6 (C=O), 142.8 (CH furílico), 140.6 (CH furílico), 125.8 (C furílico), 110.1 (CH furílico), 72.3 (CHOH), 55.2 (C), 39.3 (CH2), 36.4 (CH), 29.3 (CH2), 23.9 (CH2), 16.6 (CH3), 14.6 (CH3); Anal. C 69.85 %, H 8.56 %, calcd para C13H18O3, C 70.24 %, H 8.16 %. 4b: IR (KBr) νmáx 3443, 1692 cm-1; RMN 1H (CDCl3) δ 7.38 (1H, m), 7.35 (1H, m), 6.42 (1H, m), 4.89 (1H, sa), 2.96 (1H, sa), 2.55-2.34 (2H, m), 2.3-2.19 (1H, m), 2.03-1.71 (3H, m), 1.63-1.53 (1H, m), 0.94 (3H, s), 0.92 (3H, d, J = 7 Hz); RMN 13C (CDCl3) δ 217.1 (C=O), 142.6 (CH furílico), 140.7 (CH furílico), 125.8 (C furílico), 110.5 (CH furílico), 70.0 (CHOH), 56.6 (C), 38.9 (CH2), 38.1 (CH), 29.2 (CH2), 25.1 (CH2), 15.4 (CH3), 14.5 (CH3); Anal. C 69.96 %, H 8.29 %, calcd para C13H18O3, C 70.24 %, H 8.16 %.
Preparación de 6.
A una solución fría (0 °C) y en agitación de 0.75 g (3.38 mmol) de los cetoles 4a-4b (solos o en mezcla) en CH2Cl2 seco (10 mL), se le agregó en pequeñas porciones bajo atmósfera de argón, 1,1'-tiocarbonildiimidazol (1.5 g; 8.43 mmol). Se dejó agitando 1 h a 0 °C y después 2.5 h a t.a.. Se adicionó CH2Cl2 (20 mL) y HCl 1N (20 mL), se separó la fase orgánica y se lavó con HCl 1N (1 x 10 mL), agua (2 x 10 mL) y solución saturada de NaHCO3 (2 x 10 mL). Se secó (Na2SO4) y evaporó el disolvente obteniéndose un residuo aceitoso amarillo muy denso que se disolvió en tolueno seco (10 mL), se añadió AIBN (0.005 g) y se calentó a 75 °C bajo atmósfera de argón. Se adicionó gota a gota hidruro de tri(n-butil)estaño (1.4 g; 4.83 mmol), se mantuvo la agitación y el calentamiento durante 1 h., se enfrió, filtró y evaporó el disolvente orgánico a presión reducida y el residuo se purificó por cromatografía en columna (hexano/AcOEt, 6:1), obteniendo 0.38 g (55%) de 6 como un aceite transparente, ligero e incoloro. IR (película) νmáx 1704, 1025 cm-1; RMN 1H (CDCl3) δ 7.29 (1H, m), 7.19 (1H, m), 6.22 (1H, m), 3.01 (1H, d, J = 15 Hz), 2.42 (1H, d, J = 15 Hz), 2.36 (2H, m), 1.93-1.56 (5H, m), 1.04 (3H, s), 0.94 (3H, d, J = 7 Hz); RMN 13C (CDCl3) δ 215.4 (C=O), 142.1 (CH furílico), 141.1 (CH furílico), 120.9 (C furílico), 112.7 (CH furílico), 52.8 (C), 38.2 (CH2), 36.8 (CH), 30.51 (CH2), 29.0 (CH2), 23.9 (CH2), 19.1 (CH3), 15.3 (CH3); EMIE m/z (int. rel.): 206 [M]+ (18), 81 (100). Anal. C 75.32 %, H 9.18 %, calcd para C13H18O2, C 75.69 %, H 8.79 %.
Preparación de 7.
Bajo atmósfera de argón se adicionaron 0.4 mL (3 mmol) de cianuro de trimetilsililo a una mezcla de 6 (0.4 g; 1.94 mmol), KCN (0.02 g), éter 18-corona-6 (0.02 g) y benceno (6 mL). Se agitó a t.a. durante 1.5 h, se adicionó éter (15 mL) y se lavó con solución saturada de NaCl (3 × 10 mL). Se secó (Na2SO4) y evaporó el disolvente orgánico obteniéndose un aceite que se disolvió en THF (5 mL). A esta solución se le adicionó HCl al 10% (5 mL) y se calentó a 66 °C durante 5 h. Se enfrió, se extrajo con éter (3 × 5 mL) y la fase orgánica se lavó con solución saturada de NaCl (3 × 4 mL), se secó (Na2SO4) y evaporó el disolvente. La purificación del producto crudo por cromatografía en columna (hexano/AcOEt, 6:1) produjo 0.42 g (93%) de 7 como una mezcla diastereomérica. IR (película) νmáx 3514, 2936, 1027 cm-1; RMN 1H (CDCl3) δ 7.50 (1H, s ancho), 7.47 (1H, m), 6.45 (1H, s ancho), 2.77 (2H, s ancho), 2.63 (1H, s), 1.90-1.43 (7H, m), 1.06 (3H, s), 0.98 (3H, d, J = 6.7 Hz); RMN 13C (CDCl3) δ 144.4 (CH furílico), 141.5 (CH furílico), 121.5 (CN), 121.3 (C furílico), 111.9 (CH furílico), 77.3 (C), 44.6 (C), 38.8 (CH), 33.9 (CH2), 33.7 (CH2), 29.5 (CH2), 22.1 (CH2), 15.8 (CH3), 10.8 (CH3).
Preparación de 8.
Se adicionó POCl3 (1.5 mL) a una solución fría (0 °C) y en agitación de 7 (0.44 g; 1.88 mmol) en piridina seca (5 mL). La mezcla se dejó con agitación a 0 °C por 0.5 h, después a t.a. por 12 h y finalmente a 105 °C por 2 h. Se enfrió y se le adicionó en pequeñas porciones 15 g de hielo picado . Se extrajo con éter (4 × 8 mL) y la fase orgánica se lavó con HCl al 5% (1 × 10 mL), agua (2 × 10 mL) y solución saturada de NaCl (1 × 10 mL). Después de secar (Na2SO4) y evaporar el disolvente orgánico, se obtuvo un aceite rojizo que se purificó por cromatografía en columna (hexano/AcOEt, 12:1) produciendo 0.285 g (71%) del nitrilo insaturado 8, aceite ligeramente amarillo. IR (CHCl3) νmáx 2926, 2214 cm-1; RMN 1H (CDCl3) δ 7.33 (1H, s ancho), 7.30 (1H, s ancho), 6.60 (1H, m), 6.32 (1H, s ancho), 2.80 (1H, d, J = 15 Hz), 2.62 (1H, d, J = 15 Hz), 2.20-1.98 (2H, m), 1.71-1.46 (3H, m), 1.09 (3H, s), 0.95 (3H, d, J = 6.7 Hz); RMN 13C (CDCl3) δ 146.3 (CH furílico), 142.5 (CH furílico), 140.8 (CH vinílico), 122.0 (CN), 119.8 (C furílico), 118.9 (C vinílico), 40.2 (C), 33.0 (CH2), 31.8 (CH), 25.7 (CH2), 25.3 (CH2), 21.7 (CH3), 15.4 (CH3); EMIE m/z (int. rel.): 215 [M]+ (4), 81 (100). Anal. C 77.75 %, H 8.27 %, N 6.15 %, calcd para C14H17NO, C 78.10 %, H 7.95 %, N 6.50 %.
Preparación de 9.
Una mezcla agitada magnéticamente de 8 (0.305 g; 1.42 mmol), KOH (3.5 g), agua (8 mL) y dietilenglicol (6 mL) se calentó a la temperatura de reflujo por 72 h. Se enfrió y la solución alcalina se extrajo con éter (3 × 10 mL) para remover el material neutro. La fase acuosa se aciduló con HCl al 18%, se extrajo con éter (3 × 10 mL), la fase orgánica se lavó con solución saturada de NaCl (3 × 10 mL), se secó (Na2SO4) y se evaporó el disolvente para dar 0.09 g del ácido insaturado 9 como un aceite naranja muy espeso. La fracción neutra indicada arriba se volvió a someter al mismo tratamiento básico caliente de hidrólisis por 72 h para dar una nueva fracción ácida y otra neutra, repitiéndose finalmente el tratamiento básico caliente una vez más sobre esta última. Se obtuvo así un total de 0.24 g de 9 crudo (72% de rendimiento) el cual fue caracterizado como su éster metílico (CH2N2/éter): RMN 1H (CDCl3) δ 7.28 (1H, m), 7.1 (1H, s ancho), 6.89 (1H, m), 6.13 (1H, s ancho), 3.74 (3H, s), 3.15 (1H, d, J = 14.7 Hz), 2.59 (1H, d, J = 14.7 Hz), 2.11-2.01 (2H, m), 1.71-1.44 (3H, m), 1.20 (3H, s), 0.93 (3H, d, J = 6.7 Hz); RMN 13C (CDCl3) δ 168.1 (C=O), 142.1 (CH furílico), 141.5 (CH furílico), 140.3 (CH vinílico), 121.4 (C furílico), 112.2 (CH furílico), 51.24 (CH3), 40.1 (C), 33.5 (CH2), 31.1 (CH), 25.5 (CH2), 25.4 (CH2), 21.0 (CH3), 15.2 (CH3); EMIE m/z (int. rel.): 248 [M]+ (9), 107 (100). Anal. C 73.02 %, H 8.67 %, calcd para C15H20O3, C 72.55 %, H 8.11 %.
Preparación de 1.
Se agregó PCl5 (0.09 g; 0.43 mmol) a una solución fría (5 °C) y en agitación de 9 (0.09 g; 0.38 mmol) en benceno seco (5 mL). Después de 0.25 h la mezcla se llevó a t.a. y se mantuvo la agitación durante 1 h. Posteriormente la mezcla se enfrió a 5 °C y se adicionó gota a gota una solución de SnCl4 (0.1 mL) en benceno seco (0.5 mL). La mezcla que tomó una consistencia pastosa de color rojo-oscuro, se agitó durante 0.5 h antes de agregar consecutivamente un poco de hielo, HCl al 18% (3 mL) y éter (5 mL). Una vez separadas las fases, la acuosa se extrajo con éter (2 × 5 mL) y la fase etérea se lavó con HCl 1N (1 × 5 mL), H2O (1 × 5 mL), Na2CO3 al 5% (3 × 5 mL) y solución saturada de NaCl (2 × 5 mL). Se secó (Na2SO4) y evaporó el disolvente dando un aceite rojizo oscuro que se purificó por cromatografía en columna (hexano/AcOEt, 10:1) para dar 0.05 g (62%) de 1 como un sólido blanco, pf 150-151 °C (de éter-hexano). IR (CHCl3) νmáx 1656, 1609 cm-1; RMN 1H (CDCl3) δ 7.61 (1H, d, J = 1.7 Hz), 7.01 (1H, t, J = 4 Hz), 6.41 (1H, d, J = 1.7 Hz), 2.91 (1H, d, J = 16.5 Hz), 2.61 (1H, d, J = 16.5 Hz), 2.34-2.25 (2H, m), 1.87-1.53 (3H, m), 1.02 (3H, d, J = 6.8 Hz), 1.03 (3H, s); RMN 13C (CDCl3) δ 176.0 (C=O), 148.1 (CH furílico), 147.0 (C furílico), 142.5 (C furílico), 137.1 (CH vinílico), 136.9 (C vinílico), 112.1 (CH furílico), 40.5 (C), 39.7 (CH), 35.6 (CH2), 26.0 (CH2), 25.9 (CH2), 20.2 (CH3), 15.5 (CH3); EMIE m/z (int. rel): 216 [M]+ (100), 201 (61). Anal. C 77.42 %, H 7.17 %, calcd para C14H16O2, C 77.74 %, H 7.45 %.
Agradecimientos
Agradecemos el apoyo financiero otorgado a este trabajo por la Coordinación de la Investigación Científica de la UMSNH. Se agradece también a la Q.F.B. Concepción Armenta y al Q.F.B. José L. Salvador (UMSNH), a la Q.F.B. Rocío Patiño, a la Q. Ángeles Peña y al M.C. Javier Pérez (Instituto de Química de la UNAM) y al Departamento de Química de la UAM-Iztapalapa, por su apoyo técnico.
Referencias
1. Hikino, H.; Konno, C. Heterocycles 1976, 4, 817-870. [ Links ]
2. Torres, P.; Ayala, J; Grande, C.; Anaya, J.; Grande, M. Phytochemistry 1999, 52, 1507-1513. [ Links ]
3. Kuroyanagi, M.; Naito, H.; Noro, T.; Ueno, A.; Fukushima, S. Chem. Pharm. Bull. 1985, 33, 4792-4797. [ Links ]
4. Koike, T.; Takeuchi, N.; Ohta, T.; Tobinaga, S. Chem. Pharm. Bull. 1999, 47, 897-899. [ Links ]
5. Jacobi, P. A.; Craig, T. A.; Walker, D. G.; Arrick, B. A.; Frechette, R. F. J. Am. Chem. Soc. 1984, 106, 5585-5594. [ Links ]
6. Yamakawa, K.; Satoh, T.; Takita, S.; Iida, T.; Iwasaki, M. Chem. Pharm. Bull. 1983, 31, 3544-3552. [ Links ]
7. Irie, H.; Mizuno, Y.; Taga, T.; Osaki, K. J. Chem. Soc. Perkin Trans. 1 1982, 25-30. [ Links ]
8. Miyashita, M.; Kumazawa, T.; Yoshikoshi, A. J. Org. Chem. 1980, 45, 2945-2950. [ Links ]
9. Tada, M.; Sugimoto, Y.; Takahashi, T. Bull. Chem. Soc. Jpn. 1980, 53, 2966-2970. [ Links ]
10. Hulce, M.; Chapdelaine, M.J., in: Comprehensive Organic Synthesis, Vol. 4, Trost, B.M., Ed., Pergamon, Oxford, 1991, 237-268. [ Links ]
11. Suzuki, M.; Yanagisawa, A.; Noyori, R. J. Am. Chem. Soc. 1988, 110, 4718-4726. [ Links ]
12. Vandewalle, M.; DeClercq, P. Tetrahedron 1985, 41, 1767-1831. [ Links ]
13. Ihara, M.; Takahashi, T.; Shimizu, N.; Ishida, Y.; Sudow, I.; Fukumoto, K.; Kametani, T. J. Chem. Soc., Chem. Commun. 1987, 1467-1468. [ Links ]
14. Bohlmann, F.; Zdero, C.; Grenz, M. Chem. Ber. 1974, 107, 2730-2759. [ Links ]
15. Jakupovic, J.; Grenz, M.; Bohlmann, F.; Niemeyer, H. M. Phytochemistry 1991, 30, 2691-2693. [ Links ]
16. Warnhoff, E. W.; Martin, D. G.; Johnson, W. S. Org. Syntheses Coll. Vol. 4 1963, 162-166. [ Links ]
17. García, E.; Mendoza, V.; Guzmán, J. A.; Maldonado, L. A.; Hernández, S. Acta Cryst. 2002, C58, 336-338. [ Links ]
18. Larock, R. C. Comprehensive Organic Transformations. Wiley-VCH, New York, 1999, 44-52. [ Links ]
19. Barton, D. H. R.; McCombie, S. W. J. Chem. Soc., Perkin Trans. 1 1975, 1574-1585. [ Links ]
20. Greenlee, W. J.; Hangauer, D. G. Tetrahedron Lett. 1983, 24, 4559-4560. [ Links ]

