










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.48 no.4 Ciudad de México oct./dic. 2004
Investigación
An Unexpected Condensation Reaction Involving 2-Aminopyridine, DMF and Barbituric Acids
Héctor Salgado-Zamora,1* Ma. Elena Campos,1 Rogelio Jiménez1 and Humberto Cervantes2
1 Departamento de Química Orgánica, Escuela Nacional de Ciencias Biológicas - Instituto Politécnico Nacional. Prol. Carpio y Plan de Ayala S/N, México 11340 D.F.
2Departamento de Química, Universidad Autónoma Metropolitana-Azcapotzalco. E-mail: hsalgado47@hotmail.com
Recibido el 18 de agosto del 2004.
Aceptado el 21 de octubre del 2004.
Abstract
In this investigation the reaction of 2-aminopyridine with barbituric acid derivatives was explored. It was found that the reaction of 2-aminopyridine with 1,3-dimetylbarbituric and barbituric acids yielded an unexpected condensation product whose formation resembles a Mannich-type reaction. While the reaction with 5,5-dibromobarbituric acid yielded the corresponding 5-bromo-2-aminopyridine.
Key words: 2-aminopyridine, barbituric acid derivatives, condensation reactions.
Resumen
En esta investigación se exploró la reacción de la 2-aminopiridina con derivados de ácido barbitúrico. Se encontró que la reacción de la 2-aminopiridina con los ácidos 1,3-dimetilbarbitúrico o barbitúrico dio lugar a un producto de condensación inesperado cuya formación semeja una reacción de tipo Mannich. Mientras que la reacción con el 5,5-dibromobarbitúrico condujo a la correspondiente 5-bromo-2-aminpiridina.
Palabras clave: 2- amino piridina, derivados del ácido barbitúrico, reacciones de condensación.
Dedicated to the memory of Dr. Raymundo Cruz Almanza
Introduction
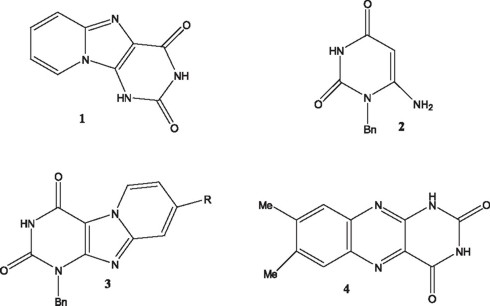
Fused tri-heterocyclic systems have attracted considerable attention due to their potential pharmacological activity, and have become valuable alternatives in drug design [1,2]. In particular we were interested in tri-heterocycles derived from purine, e.g. 1, which has a rigid, planar structure, suitable for drug evaluation.
The procedures involved in the synthesis of this type of molecules found in the literature usually require several steps [3]. Therefore it seemed interesting to investigate a direct synthetic route. A very ingenious approach that leads to an isomeric structure of 1, is that reported by Pérez-Pérez, which takes advantage of the properties associated with the uracil derivative 2. Reaction of the latter with 4-substituted pyridines, using NBS as the condensation agent, generates in one step the tricyclic system 3 [4,5]. A survey of the literature indicated that halogenated barbituric acids react with o-phenylen diamine derivatives in a general fashion to yield alloxazines [6], e.g. 4 (from 4,5-dimethyl-o-phenylendiamine). Also, pyrimidinothiazoles have been synthesized through a condensation reaction involving thioacetamide and bromodimethylbarbituric acid [7].
Results and Discussion
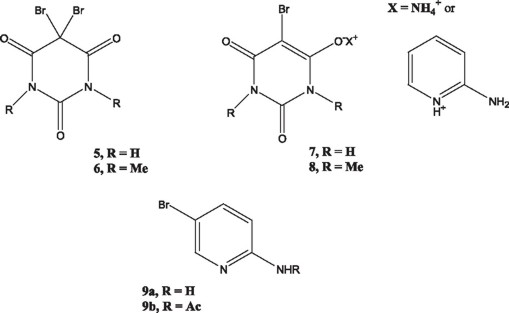
The synthetic approach involving dibromobarbituric acids was attractive. It was envisioned that 2-aminopyridine might be utilized instead of the o-phenylendiamine in the reaction with 5,5-dibromobarbituric acids to give the desired 1. Accordingly, 5,5-dibromobarbituric acid (DBBA, 5) was prepared from barbituric acid and an excess of bromine in water following a literature procedure [6]. Similarly, 1,3-dimethyl-5,5-dibromobarbituric acid (DMDBBA, 6) was prepared from 1,3-dimethylbarbituric acid and bromine (2 moles) in acetic acid or with NBS (2 moles) in DMF to avoid the employment of the corrosive and irritating bromine. Both brominated products were identified through their melting points, which were similar to those reported in the literature [8].
The reaction of 2-aminopyridine (2-AP) with 5,5-dibromobarbituric acid was performed following a related literature protocol [9]. Accordingly, an excess of 2-AP (1.7 g) was heated to its melting point temperature (ca. 60 °C) and at this point DBBA (0.15 g) was added. The reaction mixture turned deep brown above 100 °C and was kept at 125 - 130 °C for 3 h. Thin layer chromatography showed that all starting DBBA had been consumed, however after the usual work up of the reaction crude no product was isolated in the organic solvent even after pH variations. Water removal from the aqueous phase left a solid residue, which was identified as the salt 7 (X = 2-aminopyridinium), a non-surprising result. The experiment was repeated in various solvents using only 2 equivalents of 2-AP. In DMF, the main product was again the salt and in dioxane, the reaction was incomplete and afforded a complex mixture of products. In acetic acid, traces of compound 1 could be detected.
The reaction of 2-AP with the dibromo derivative 6 was then performed in acetic acid and in ethyl alcohol. In the latter solvent, a non-polar product observed in the reaction mixture was isolated and identified as 2-amino-5-bromopyridine 9a. In acetic acid, another product with a very similar rf as the starting dibromo compound 6 was detected. Purification of the crude mixture by column chromatography afforded a white crystalline solid, which was shown to be the acylated 2-amino-5-bromopyridine 9b, clearly identified by both lH NMR and 13C NMR spectra1 data. The result might appear unimportant if it were not for the fact that bromination of 2-aminopyridine usually results in the mixture of 3- and 5-bromo-2-aminopyrydine or 3,5-dibromo-2-aminopyridine [10]. Thus, this dibromo-1,3-dimethylbarbituric acid might find useful applications as a mild brominating agent.
In view of the above described results, the corresponding monobromo ammonium salts 7 and 8 (X = NH4+ ) were prepared [5] and submitted to reaction with 2-AP (only one mole) in glacia1 acetic acid. However, in both cases the main product was again the 2-aminopyridinium sa1t. 2-Acylaminopyridine could also be detected.
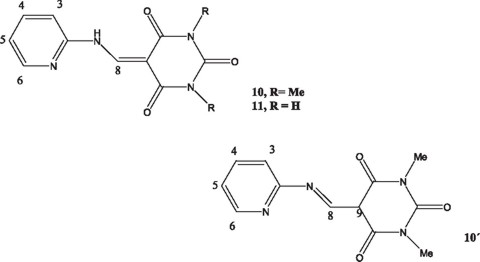
Then, the possibility of reacting 2 equivalents of 2-AP (2 moles) with the barbituric acids was considered. It was assumed that one mole pf 2-AP would be consumed in the typical acid-base reaction and a second mole would add to the double bond formed to give a precursor of product 1. Thus, 2-AP was treated with 1,3-dimethylbarbituric acid in DMF at 70 °C (heating above this temperature led to decomposition). The reaction gave recovered starting materials and a white solid for which structure 10, has been assigned on the basis of the following spectral data: The mass spectrum of the compound gave a molecular ion at m/z 260. The 1H NMR spectrum of the isolated compound (Fig. 1) showed 6 hydrogens in the aromatic region, which were accounted for as follows: the doublet at δ. 9.38 was assigned to H-6, the multiplet at 8.40 to H-8, the triplet at δ 7.65 was assigned to H-5, whereas the up field doublet (6.96 ppm) was designated to H-3. The multiplet at δ 7.05 was assigned to H-4. The two methyl groups were considered magnetic equivalent and explain the singlet observed at δ 3.3. The 13C NMR spectrum of the product confirmed the existence of 10 carbon atoms, in addition of the two methyl signals in the up field region. The floppy physical consistency of the product did not allow an X-ray analysis. The isomeric structure 10' was ruled out since no signal in the 13 C NMR spectrum was detected for carbon C-9 (CH). As expected, the reaction carried out with barbituric acid under similar experimental conditions afforded structure 11.
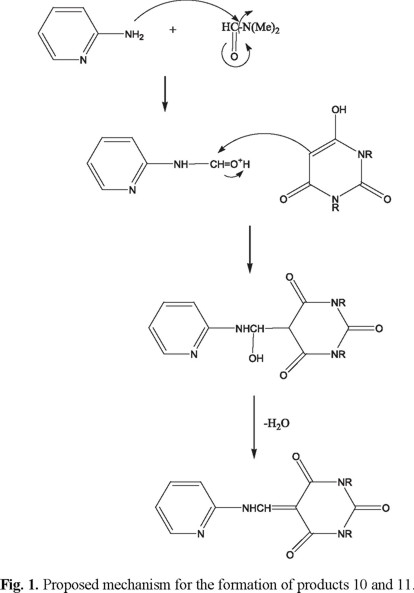
A possible mechanism for the formation of the product is depicted in Figure 1 and involves a transamidation reaction of 2-AP and DMF catalyzed with barbituric acid as the first step. Then the enol of barbituric acid reacts with the newly formed amide, to yield the observed product after losing a molecule of water. Overall, the process is reminiscent of a Mannich-type reaction
Conclusion
It was concluded that the reaction of 2-AP with dibromobarbituric acids affords as the main product, the salt formed after one of the bromine atoms has been displaced. 5,5-Dibromo-1,3-dimethylbarbituric acid turned out to be a mild brominating agent suitable for activated molecules. The reaction in DMF of 2-aminopyridine with barbituric acid or 1,3-dimethylbarbituric acid led respectively to an unexpected condensation product, possibly formed through a path resembling a Mannich-type process. Other amines and nitrogen nucleophiles are currently under investigation.
Experimental
Melting point were measured on an Electrothermal melting point apparatus and are uncorrected. 1H and 13C NMR spectral data were recorded at 300 and 75 MHz respectively using a Mercury 300 MHz NMR spectrometer. Chemical shifts (δ) are given in parts per million downfield from TMS (δH=0). Mass spectra were obtained with a Jeol JMS AX505HA instrument. Column chromatography was carried out with silica gel (Merck 60, 70-230 mesh) as the adsorbent. All reactions were carried out under nitrogen atmosphere.
2-Amino-5-bromopyridine (9a). 2-Aminopyridine (0.3 g. 3.18 mmol) was dissolved in EtOH (10 mL). To this solution, 5,5-dibromobarbituric acid (0.5 g, 1.6 mmol) in ethyl alcohol (10 mL)was added. The formed suspension dissolved upon heating (70 ºC) and was left on stirring overnight at this temperature. Then the reaction mixture was allowed to cool to room temperature, filtered, EtOH removed under vacuum, water was added and the residue extracted with CH2Cl2 (3 × 15 mL). Extracts were combined, dried (anh. Na2SO4) and solvent removed under reduced pressure to leave a residue which crystallized upon addition of toluene, to afford 300 mg ( 83.8 %) of the title compound as a white solid, mp 134-136 ºC (Lit.[9] mp 132 - 135 ºC).
1,3-Dimethyl-5-methylidene(2-aminopyridyl)barbituric acid (10). 2-Aminopyridine (1.47 g, 15.6 mmol), and 1,3-dimethylbarbituric acid (1.22 g, 7.8 mmol) were dissolved in DMF (25 mL) and heated for 4 h at 70 ºC. Then the reaction mixture was cooled, poured into iced water and extracted with EtOAc (3 × 15 mL). Extracts were combined and dried (anh. Na2SO4). Solvent was removed under reduced pressure to leave a residue which crystallized upon addition of ethyl alcohol (5 mL). The solid was collected by filtration to give 350 mg (17.24 %) of the title compound as white floppy plates, mp 200 - 201 ºC. 1H NMR (CDCl3) δ 12.0 (m, 1H), 9.38 (d, J = 9 Hz, 1H), 8.40 (m, 1H), 7.85 (t, J = 5Hz, 1H), 7.05 (m, 1H), 6.96 (d, J = 5 Hz, 1H), 3.26 (s, 6H). 13C NMR δ 26.2, 27.8, 94, 113, 121, 139, 148.5, 148.7, 151.4, 151.8, 162.3, 164.5. MS m/z 260 (M+), 79 (100%). Anal. Calcd. for C12H12N4O3: C, 55.38; H, 4.61; N, 21.53. Found: C, 55.30; H, 5.2; N, 21.48.
5-Methylidene(2-aminopyridyl)barbituric acid (11). Similarly, 2-aminopyridine (1.47 g, 15.6 mmol) and barbituric acid (1.0 g, 7.8 mmol) reacted to give 310 mg (17.1 %) of the title compound as white plates, mp 253.5 - 254.5 ºC. 1H NMR (DMSO)d6 δ 11.0 (m, 1H), 8.38 (dd, J = 1 Hz, J = 4 Hz, 1H), 8.25 (s, 1H), 7.98 (dd, J = 1 Hz, J = 5 Hz, 1H), 7.82 (dd, J = 5 Hz, 1H), 7.20 (dd, J = 1 Hz, J = 4 Hz, 1H), 3.1-3.5 (m, 2H). 13C NMR δ 110, 121, 129, 137, 144.5, 146, 150, 153.6, 160.2, 161.5. MS 232 (M+), 79 (100 %). Anal. Calcd. for C10H8N4O3: C, 51.72; H, 3.44; N, 24.13. Found: C, 51.78; H, 3.38; N, 23.60
Acknowledgement
Financial aid from I.P.N. México, through project CGPI 20030744, is gratefully acknowledged.
References
1. Maitre, M.; Hechler, V.; Vayer, P.; Gobaille, S.; Cash, C. D.; Schmitt; Bourguignon, J. J. Pharmacol. Exp. Ther. 1990, 255, 657-660. [ Links ]
2. Salgado-Zamora, H.; Rizo, B.; Campos, E.; Jiménez, R.; Reyes, A. J. Heterocyclic Chem. 2004, 41, 91 - 94. [ Links ]
3. Hamton, A.; Harper, P. J.; Sasaki, T. Biochemistry 1972, 11, 4965-4969. [ Links ]
4. Priego, E. M., Kuenzel, J.; Ijzerman, A. P.; Camarasa, M. J.; Pérez-Pérez, M-J. J. Med. Chem. 2002, 45, 3337-3344. [ Links ]
5. Pérez-Pérez, M. J.; Priego, E. M.; Jimeno, M. N.; Camarasa, M-J. Organic Lett. 2002, 4, 155-157. [ Links ]
6. Tishler, M.; Wellman, J. W.; Ladenburg, K. J. Am. Chem. Soc. 1945, 67, 2165-2168. [ Links ]
7. Erlenmeyer, H.; Furger, H.P. Helv. Chim. Acta. 1947, 30, 585-592. [ Links ]
8. Biltz, H.; Hamburger, T.; Ber. 1916, 49, 635-652. [ Links ]
9. Taylor, E. C.; Paudler, W. W.; Cain, C. K. J. Org. Chem. 1955, 20, 264-270. [ Links ]
10 Fox, B.A.; Threlfall, T. L. Org. Synth. Coll. Vol. V. pp 346 - 351. John Wiley & Sons, New York, NY, 1973. [ Links ]

