










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.46 no.4 Ciudad de México oct./dic. 2002
Investigación
Reduction Potentials of Conjugated Aliphatic Ketones, Oximes, and Imines: Correlation with Structure and Bioactivity
Noah N. Niufar,1 Fiona L. Haycock,1 Jodi L. Wesemann,1 Jason A. MacStay,2 Victor L. Heasley*2 and Peter Kovacic3
1 Department of Chemistry, Saint Mary's College of California, Moraga, CA 94575, USA.
2 Department of Chemistry, Point Loma Nazarene University, San Diego CA 92106, USA. Tel: 619-849-2231; Fax: 619-849-2598. E-mail: vheasley@ptloma.edu)
3 Department of Chemistry, San Diego State University, San Diego, CA 92182, USA.
Recibido el 20 de septiembre del 2002.
Aceptado el 12 de noviembre del 2002.
Abstract
To determine which structural features may favor electron transfer in biological systems, cyclic voltammetric studies were carried out on conjugated aliphatic ketone, oxime, and imine species at pH levels 2 through 7 in most cases. The nature of the conjugated substituents strongly influenced the reduction potentials. The dione samples were most easily reduced, followed by the diimine and then the mono- and di-oxime analogs. The reduction potentials for all compounds were pH-dependent, with the most favorable potentials occurring at the lower pH values. The possible roles of protonation, hydrogen bonding, the captodative effect, and other aspects were considered. The favorable reduction potentials of the compounds ex-hibiting bioactivity suggest that in vivo electron transfer and oxidative stress may be involved in various types of biological processes.
Keywords: Cyclic voltammetry; diones; diimines; dioximes; bioactive; electron transfer; oxidative stress.
Resumen
Para determinar las características estructurales que pueden favorecer la transferencia electrónica en sistemas biológicos, se llevaron a cabo estudios de voltametría cíclica en cetonas conjugadas, oximas e iminas a pH de 2 a 7. La naturaleza de los substituyentes conjugados tienen notable influencia en los potenciales de reducción. Las dionas se reducen más fácilmente, seguido por las diiminas y los análogos mono- y di- oximas. Los potenciales de reducción de todos los compuestos son dependientes del pH, y las potencias más favorables se encuentran a pH bajos. Los posibles efectos de protonación, formación de puente de hidrógeno, efecto captodativo, y otros aspectos, también fueron considerados. Los potenciales de reducción favorables de los compuestos que muestran actividades biológicas sugieren que la transferencia electrónica in vivo y el estrés oxidativo pueden estar involucrados en varios tipos de procesos biológicos.
Palabras clave: Voltametría cíclica, dionas, diiminas, dioximas, bioactividad, transferencia electrónica, estrés oxidativo.
Introduction
During the past 40 years, a large volume of research has given support for the involvement of oxidative stress (OS) and electron transfer (ET) in the mode of action of physiologically-active compounds, such as anti-infective drugs [1], anticancer drugs [2], carcinogens [3], reproductive toxins [4], and ne-phrotoxins [5], many of which contain ET functionalities. Several classes of ET agents are quinones (phenolic precursors), metal complexes (complexors), aromatic nitro compounds (reduced derivatives), and conjugated imines (iminium species). These principal types can participate in redox cycling with O2 to generate various reactive oxygen species, e.g., H2O2, ROOH, ROOR, O.-, HO., ROO., which can bring about beneficial effects, as in drug action, or alternatively, they can cause unwanted toxic manifestations from body cell insult. ET agents can also interfere with normal electron transport processes taking place in the central nervous system or the respiratory chain. Thus, the ET groups generally present in bioactive substances or their metabolites presumably play a functional role.
Electrochemistry has been increasingly helpful in elucidating the basic chemistry of biological systems. Substances with reduction potentials above -0.5 V (SHE) may undergo ET in vivo [6]. Of relevance are the eletrochemical studies of the conjugated imine and iminium moieties found in the following biological agents: an oxidative metabolite of the antimalarial primaquine [7], the anti-infective gentian violet [8], the anti-infective iminodaunorubicin [9], a metabolite of anti-infective ethionamide [10], the anticonvulsant progabide [11], a nicotine metabolite [12], a metabolite of phencyclidine [13], the sedative benzodiazepines [14], and a metabolite of anticancer a-difluoromethylornithine [15]. Electrochemical investigations of bioactive conjugated oximes have also been done on the amebicidal oxime of entobex [9], the oxime of antifungal griseofulvin [8], antibiotic monolactams [16], and the antiviral zinviroxime [17]. In comparison α-dicarbonyl species have received little attention. Conjugated α-dicarbonyls (o-quinones) are represented by the antibacterial bonaphthon [18], the amebicidal entobex [10], and the oxidative metabolite of the antimalarial primaquine [7]. Although direct correlations between reduction potentials and biological activity can be difficult to establish, due to the important roles played by other factors, such as solubility, metabolism, diffusion, absorption, cell permeability, site binding, and stereochemistry, evidence indicating that ET plays a role in biological processes has been extensive [1-19]. See the discussion for bioactivity of materials in the present study.
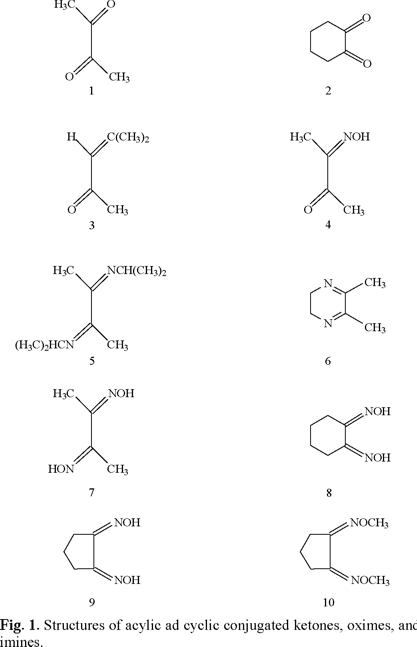
One research aim was to determine reduction potentials of simple aliphatic α-diketones and related oxime and imine derivatives at 2-7 pH. Correlations are made between reduction potentials and the aspects of protonation, H-bonding, the captodative effect, and physiological activity. Metabolism may play a role since ketoximes are oxidatively converted in vivo to nitric oxide [20] which displays a wide range of bioactivity. ET, as previously discussed for related biologically active compounds [1-19], may participate in vivo as part of a multifaceted scenario.
It should be noted that this study addresses general trends. Since ET agents act catalytically, we are not concerned with number of electrons taken up or products of in vitro reactions. Representative references are used in some cases, and background literature is not exhaustively reviewed.
Experimental
1. Materials
Potassium chloride, sodium phosphate, and citric acid were acquired from Fluka. The samples of 2,3-butanedione (1), 1,2-cyclohexanedione (2), mesityl oxide (3), 2,3-butanedione monoxime (4), 1,2 cyclohexanedione dioxime (8), and acetone were acquired from Aldrich in 97 % purity or higher. Literature preparations were followed for 2,3-di-n-propyliminobutane (5) [21], 2,3-dihydro-4,5-dimethylpyrazine (6) [22], and 1,2-cyclopentanedione dioxime(9) [23]. Compound (6), unstable in air at room temperature, should be kept in the cold under nitrogen.
2. Synthesis of 1,2-cyclopentanedione dimethyloxime (10)
To a stirred solution of 106 mg (1.08 mmol) of 1,2-cyclopentadione and 1.5 mL of pyridine in a round-bottom flask was added 108 mg (2.15 mmol) of methoxyamine hydrochloride. After the reaction proceeded for 30 minutes, the pyridine was removed under vacuum and the viscous liquid was distilled under vacuum (74 °C/2 Torr) to give white crystals that melted at 43-46 °C. The structure of 10 was established by its mass spectrum (MS) and nuclear magnetic resonance spectrum (NMR). MS: GC-MS, m/z, relative intensity (%) (EI): 156 (M, 30), 125 (M-OCH3, 4.7) 95 (M-OCH3-CH2O, 100). 1H NMR (60MHz): δ 1.86 (quintet, 4H), 2.61 (t, 2H), 3.90 (s, 6H).
3. Cyclic voltammetry
Solutions of 2.0 mM concentration were prepared in deionized water using McIlvaine buffers (potassium chloride, sodium phosphate, and citric acid) with an ionic strength of 0.5 M [24]. The solutes were dissolved using ultrasound for a period of five to ten minutes. Compounds in acidic solution dissolved most readily, with the cyclic forms doing so faster than acyclic forms. A Unifet hand-held pH meter Model UF100 calibrated with buffers of pH 4 and 7 was used to measure the pH of all solutions which were run immediately after their preparation. Approximately 10 mL of each sample was poured into the CV cell and purged with nitrogen gas for fifteen minutes.
The EG&G model 270 Research Electrochemistry software was used to control an EG&G Model 263 Potentiostat and EG&G model 303 SMDE. The cyclic voltammograms were run with a hanging mercury drop electrode of triply distilled mercury. An Ag / AgCl and platinum wire were used as the reference and counter electrodes, respectively. A rate of 50 mV / s was used to scan the region between 0 and -2.2 V vs the Ag / AgCl electrode. The reduction potentials reported here are averages of the cathodic potentials (Ec), converted to SHE by adding 0.22 V.
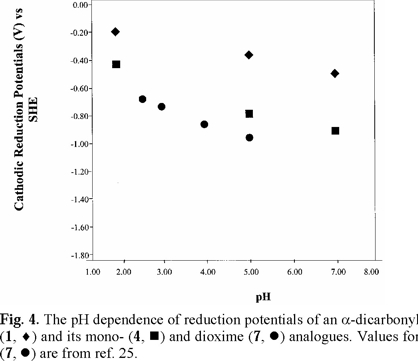
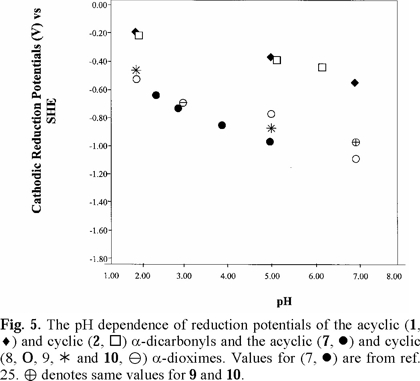
For 2,3-butanedione dioxime (dimethylglyoxime) (7), literature polarographic data [25] were plotted in Figs. 4 and 5 (converted to SHE). Values for pH 5.98 and 7.03 were not used because of uncertainty.
Results and discussion
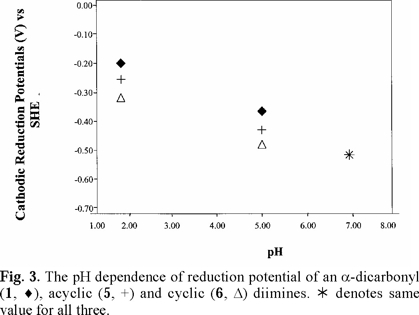
Cathodic reduction potentials for 1-10 at various pH values are shown in Figs. 2 through 5 (3, 4). All reactions occurred between -0.2 and -1.2 V vs. SHE. To increase the validity of comparison, studies were performed in the same solvent and electrochemical system. Use of McIlvaine buffers allowed the same buffering species and ionic strength to pertain over the entire pH range.
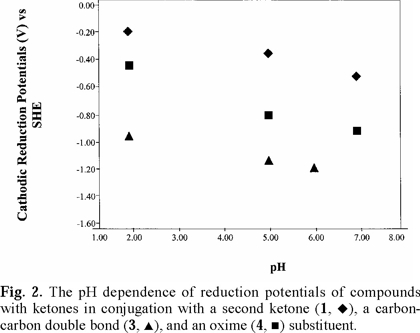
Since none of the compounds exhibited reversible redox reactions, potentials of the cathodic peaks are reported as Ec, rather than E1/2. The compounds 1, 5 and 6 gave irreversible oxidation peaks on the return sweep, while all others had only reduction peaks. Lack of reversibility indicates that, under these conditions, the species is reduced, reacts further, and thus cannot be returned to the original state. In vivo, however, the reaction may be reversible due to relative isolation of the compound at the binding site.
1. pH dependence, protonation and hydrogen bonding
Studies were done over the pH range of 2-7, encompassing some values that are found in the human body and hence have physiological relevance. Figs. 2 through 5 (3, 4) show that the Ec values are pH-dependent, with reduction of all species occurring most easily at lower pH where protons are readily available. Most of the dependencies are not linear over the entire range, suggesting that various mechanisms occur at different pH values.
The roles that protons play in the processes depend on the nature of the species being reduced, particularly their basicity. More basic imines and oximes are likely to be protonated prior to reduction. Literature eletrochemical data established that the protonated oxime is the reducible species in acidic solution [26]. Less basic species will remain unprotonated until after reduction occurs. Prior studies on 2 show that at pH values below 10, initial one-electron reduction is followed by protonation [27].
Protonation of the bases prior to reduction is obviously playing a role of some type. Since electrons are attracted to positively-charged species, protonation should favor reduction, as was seen in previous studies of imines and oximes [1-19]. However, the relative effect of pH observed for the easily-protonated imines in these studies was not as great as that for the dicarbonyl species. At about neutral pH, dicarbonyl 1 and diimine 5 are reduced with roughly equal ease. At lower pH, where the diimine is more likely to be protonated, reduction of 5 occurs less readily. A similar situation pertained to 6, which is a cyclic diimine analog of 5 (Fig. 3), and to Fig. 4 in which ease of electron uptake is in the order of 1 > 4 > 7, for the entire pH range. A similar situation pertained to the cyclo-hexane analogs 2 and 8 (Fig. 5).
In marked contrast, studies performed under acidic conditions demonstrated that acetophenone oxime [16] is much more readily reduced that the parent ketone [28].
Earlier studies convincingly illustrate the important impact of hydrogen bonding. Positive shifts in the reduction potentials for quinones were attributed to stabilization of the reduced radical anions by hydrogen-bonding [29]. In studies of o-quinones (α-dicarbonyls) in the presence of substituted ureas, large positive shifts in redox potential occurred, which apparently resulted from stabilization of the radical anion by hydrogen-bonding [30]. The effect was greatest when the two hydrogens of the ureas interacted simultaneously with the reduced quinone. Also, complexes of flavins (conjugated diimines) with hydrogen-bonding receptors showed more po-sitive reduction potentials than the parents, indicating substantial stabilization of the radical anions [31]. Since hydrogen bonds can be formed by all the species studied here, the reduction potentials may be influenced, at least in part, by these interactions.
2. Role of conjugated substituents
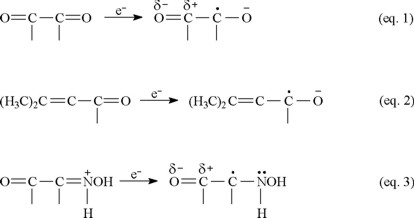
Conjugation plays an important role in favoring reduction of ketones, stabilizing the one-electron reduction products. Compared to acetone, whose reduction potential does not fall in the range of the McIlvaine buffer, 1 is easily reduced. The electron gained by 1 can be delocalized over the conjugated system, thereby stabilizing the radical anion (Equation 1).
The captodative effect, the enhanced stabilization that occurs when a carbon radical is attached to both an electron-withdrawing and an electron-donating substituent [32], is believed to play a role in physiological activity. Pertinent examples from biological systems [33] are paraquat, flavin enzymes, quinone-containing enzymes and drugs, and the ring-opened imine carboxylic acid formed from enzyme attack of the β-lac-tam moiety of cephalosporins. The greater ease with which 1 is reduced compared to 3 (Fig. 2) can be attributed in part to the captodative effect. The one-electron reduction product in equation 1 is stabilized by the electron-withdrawing carbonyl group and the negatively-charged oxygen, incorporating resonance delocalization. In contrast, the one-electron reduction product in equation 2 does not have the benefit of an electron-withdrawing group and is not as easily formed. Equation 3 illustrates the captodative effect applied to 4 in protonated form.
3. Configuration and conformation (cyclic substrates)
In general, acyclic compounds favor the transoid form over cisoid because of steric hindrance. However, hydrogen-bonding, which can occur in the oximes and protonated diimines, could cause the species to spend time in the cisoid arrangement. In the cyclic compounds, the cis arrangement is locked into place.
Previous studies on various α-dicarbonyl derivatives indicate that reduction is less favored the more time that the acyclic compounds spend in a transoid arrangement. Progressive substitution of alkyl groups in the α, α'-positions of 1 resulted in increasingly negative reduction potentials [34]. The results were attributed to steric hindrance, to transcoplanarity of the dicarbonyl system, and to 'insulation' from the electrode. Polarographic differences are known to exist between syn- and anti-oximes [35].
Fig. 2-5 (3, 4) incorporate data for both acyclic and cyclic substrates. Such reasoning [34] apparently does not apply in a number of our cases. In comparisons of reduction potentials of acyclic and cyclic categories, acyclic 1 and cyclic 2 were approximately equal (Fig. 5); acyclic 7 was more negative than cyclic 8 which was similar to cyclic 9 (Fig. 5); whereas cyclic 6 was more negative than acyclic 5 except for equality at pH 7. Compound 10, in which methyls replaced oxime hydrogens, was essentially equal to 9 in ease of reduction (Fig. 5). Other mechanistic factors to consider are electron-donating inductive effect of alkyl groups, repulsive influence of the negative charges on the oxygens and nitrogens of the cyclic radical anions, and configuration of the oximes.
4. General mechanistic considerations
We deem it prudent to limit speculation. A detailed discussion of mechanism becomes quite uncertain due to complexity involving a variety of influences, such as, electronegativity, resonance, inductive effect, steric factors, captodative influences, protonation, H-bonding, configuration, conformation, hydration, and solvation. Hydration, which occurs in some cases, eliminates conjugation, and hence drastically reduces ease of electron uptake. Regarding mechanisms of physiological activity, in addition to ET, a number of other factors are likely operating including metabolism (vide infra).
5. Correlation with bioactivities
All of the principal compounds studied, except 7, gave reduction potentials in the physiologically active range. As noted above, establishing direct correlations between reduction potentials and biological activity is challenging. However, the fact that 1, 2 and 4, the compounds that were readily reduced, have already been shown to have biological activity suggests that ET may be playing a role.
A number of α-dicarbonyl compounds exhibit mutagenic properties [36, 37]. These include 1, which also induces sister-chromatid exchange [38], and 2, which is also an antiviral agent [39]. It is intriguing that 1, a simple α-diketone, is electron affinic [40] and can condense in vivo with protein amino groups to form conjugated imine or iminium species. The resulting mono-imines would be analogous to the bioactive monoxime derivative (4). Little recognition has been given to a-dicarbonyls, other than o-quinones, as potential ET agents.
The physiological responses of 4 have been the object of much study, with most attention being devoted to the effects on cardiac muscle tension, neuromuscular transmission, action potential, and ion currents [41]. Although these phenomena have been attributed to dephosphorylation of substrates and direct channel blocks, we suggest the possibility of ET partici-pation. This compound also displays some activity as an anti-dote for nerve gas, whose action has been attributed, in part, to ET [42].
A recent study was carried out on the metabolism of acetone oxime [20]. Oxidation by cytochrome P450 generated nitric oxide via a radical pathway in which, apparently, hydroxyl-type radicals play a role. An earlier review presented evidence for OS-ET in the various bioactivities of NO [43]. Much attention has been devoted to involvement of peroxynitrite, formed by interaction of NO with superoxide, in physiological activity. As is often the case with drugs, a multifaceted approach may be applicable to the biological action of the various substrates.
In conclusion, all three types of conjugated substrates (ketones, oximes, and imines) gave reduction potentials conducive to ET in vivo. Mechanistic aspects of electroreduction are discussed entailing correlation with structure. Various bioactivities may involve ET or NO production from oximes.
Acknowledgements
This research was sponsored by Abbott Laboratories, Genentech, the School of Science at Saint Mary's College of California, the Research Associates of Point Loma Nazarene University, and the SDSU-MIRT Program under the direction of Dr. R.S. Pozos, NIH No. 5T37TW00067. We thank Professor Diane Smith (SDSU) for helpful discussions.
References
1. Kovacic, P.; Becvar, L. E. Curr. Pharm. Des. 2000, 6, 143-167. [ Links ]
2. Kovacic, P.; Osuna, Jr. P. E. Curr. Pharm. Des. 2000, 6, 277-309. [ Links ]
3. Kovacic, P.; Jacintho J. D. Curr. Med. Chem. 2001, 8, 773-796. [ Links ]
4. Kovacic, P.; Jacintho, J. D. Curr. Med. Chem. 2001, 8, 863-892. [ Links ]
5. Kovacic, P.; Sacman, A.; Wu-Weis, M. Curr. Med. Chem. 2002, 9, 849-867. [ Links ]
6. Frank, D. M.; Arora, P. K.; Blumer, J. L.; Sayre L. M. Biochem. Biophys. Res. Commun. 1987, 147, 1095-1104. [ Links ]
7. Ames, J. R.; Ryan, M. D.; Klayman, D. L.; Kovacic, P. Free Rad. Biol. Med. 1985, 1, 353-361. [ Links ]
8. Kovacic, P.; Kiser, P. F.; Feinberg, B. A. Pharm. Res. 1990, 7, 283-288. [ Links ]
9. Ames, J. R.; Hollstein, U.; Gagneux, A. R.; Ryan, M. D.; Kovacic, P. Free Rad. Biol. Med. 1987, 3, 85-96. [ Links ]
10. Kovacic, P.; Ames, J. R.; Ryan, M. D. Bioelectrochem. Bioenerg. 1989, 21, 269-278. [ Links ]
11. Ames, J. R.; Kovacic, P.; Kadaba, P. K.; Kiser, P. F. Epilepsia 1992, 33, 936-943. [ Links ]
12. Kovacic, P.; Kassel, M. A.; Castonguay, A.; Kem, W. R.; Feinberg, B. A. Free Rad. Res. Commun. 1990, 10, 185-192. [ Links ]
13. Kirschner, K. N.; Van Dyke, C.; Kovacic, P.; Bowen, J. P. THEOCHEM 2000, 498, 167-179. [ Links ]
14. Crawford, P. W.; Kovacic, P.; Gilman, N. W.; Ryan, M. D. Bioelectrochem. Bioenerg. 1986, 16, 407-426. [ Links ]
15. Kovacic, P.; Ames, J. R.; Taylor, E. C.; Ryan, M. D. J. Pharm. Sci. 1988, 77, 999-1002. [ Links ]
16. Kovacic, P.; Ames, J. R.; Ryan, M. D. Bioorg. Chem. 1988, 16, 149-164. [ Links ]
17. Kovacic, P.; Kassel, M. A.; Popp, W. J.; Feinberg, B. A. J. Biopharm. Sci. 1990, 1, 315-329. [ Links ]
18. Ames, J. R.; Ryan, M. D.; Kovacic, P.; J. Free Rad. Biol. Med. 1986, 2, 377-391. [ Links ]
19. Kovacic, P.; Jawdosiuk, M.; Ames, J. R.; Ryan, M. D. Bioorg. Chem. 1987, 15, 432-441. [ Links ]
20. Stoyanovsky, D. O. Nitric Oxide 2001, 5, 413-424. [ Links ]
21. Dieck, H. T.; Franz, K. D.; Majunke, W. Z. Naturforsch 30B, 922-925. [ Links ]
22. Flament I.; Stoll M. Helv. Chim. Acta 1967, 50, 1754-1758. [ Links ]
23. Bassett, J.; Leton, G. B.; Vogel, A. I. Analyst 1967, 92, 279-289. [ Links ]
24. Elving, P. J.; Markowitz, J. M.; Rosenthal, I. J. Electroanal. Chem. 1956, 28, 1179-1180. [ Links ]
25. Spritzer, M.; Meites, L. Anal. Chim. Acta 1962, 26, 58-65. [ Links ]
26. Lund H. Acta Chem. Scand. 1959, 13, 249-267. [ Links ]
27. Zon, M. A.; Rodriguez, Mellado, J. M. J. Electroanal. Chem. 1992, 338, 229-238. [ Links ]
28. Bard, A. J. ed. Encyclopedia of Electrochemistry of the Elements. New York: Marcel Dekker 1978, Vol. 12, pp. 54-55. [ Links ]
29. Gupta, N.; Linschitz, H. J. Am. Chem. Soc. 1997, 119, 6384-6391. [ Links ]
30. Ge, Y.; Miller, L.; Ouimet, T.; Smith, D. K. J. Org. Chem. 2000, 65, 8831-8838. [ Links ]
31. Breinlinger, E.; Rotello, V. M. J. Am. Chem. Soc. 1997, 119, 1165-1166. [ Links ]
32. Viehe, H. G.; Janousek, Z.; Merenyi, R.; Stella, L. Acc. Chem. Res. 1985, 18, 148-154. [ Links ]
33. Kovacic, P.; Popp, W. J.; Timberlake, J. W.; Ryan, M. D. Chem. Biol. Interact. 1989, 69, 235-244. [ Links ]
34. Leonard, N. J.; Laitinen, H. A.; Mottus, E. H. J. Am. Chem. Soc. 1953, 75, 3300-3303. [ Links ]
35. Bard, A. J. ed. Encyclopedia of Electrochemistry of the Elements. New York: Marcel Dekker, 1978, Vol. 13, pp. 318-322. [ Links ]
36. Bjeldanes, L. F.; Chew, H. Mutat. Res. 1979, 67, 367-371. [ Links ]
37. Nehlig, A.; Debry, G. Mutat. Res. 1994, 317, 145-162. [ Links ]
38. Tucker, J. D.; Taylor, R. T.; Christensen, M. L.; Strout, C. L.; Hanna, M. L.; Carrano, A. V. Mutat. Res. 1989, 224, 269-279. [ Links ]
39. Tiffany, B. D.; Wright, J. B.; Moffett, R. B.; Heinzelman, R. V.; Strube, R. E.; Aspergren, B. D.; Lincoln, E. H.; White, J. L. J. Am. Chem. Soc. 1957, 79, 1682-1687. [ Links ]
40. Jacobs, G. P. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1986, 49, 887-890. [ Links ]
41. Schlichter, L. C.; Pahapill, P. A.; Chung, I. J. Pharmacol. Exp. Ther. 1992, 261, 438-446. [ Links ]
42. Kovacic, P. Curr. Med. Chem. in press. [ Links ]
43. Kovacic, P. Bioelectrochem. Bioenerg. 1996, 39, 155-159. [ Links ]

