










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.46 no.2 Ciudad de México abr./jun. 2002
Investigación
Evidence for a Strained Cyclic Allene in the Ring Closure Reaction of Enynylketenes
Mario Fernández,*1 Alejandro Ramírez,2 Ramón Hernández,1 Mario Ordóñez1
1 Centro de Investigaciones Químicas, Universidad Autónoma del Estado de Morelos, Av. Universidad 1001. Chamilpa, Cuernavaca 62210, Morelos, México. Tel/Fax: 01(777) 329-7997. E-mail: mfz@ciq.uaem.mx
2 Facultad de Ciencias, Universidad Autónoma del Estado de Morelos.
Recibido el 20 de febrero del 2002.
Aceptado el 19 de junio del 2002.
Abstract
We show in this paper using HF and MP2 ab initio calculations that the ring closure of 5 generates an intermediate best described as the closed-shell cyclic allene 6a, instead of the usually assumed diradical nature reported in the literature. This intermediate then gives rise to the formation of 7. The closed-shell cyclic allene nature of the lowest-lying singlet was confirmed using CASSCF calculations.
Keywords: cyclic allene, Hartree-Fock, MP2, CASSCF, benzoquinones.
Resúmen
Utilizando cálculos HF y MP2 ab initio, demostramos que la reacción de ciclización de 5 genera un intermediario cuyas características concuerdan con las de un aleno cíclico de capa cerrada 6a en lugar de la usualmente aceptada naturaleza diradicálica reportada en la literatura. Este intermediario, posteriormente da lugar a la formación de las benzoquinonas 7. La naturaleza de aleno cíclico de capa cerrada fue confirmada con cálculos CASSCF.
Palabras clave: aleno cíclico, Hartree-Fock, MP2, CASSCF, benzoquinonas.
Introduction
Strained cyclic allenes have recieved considerable attention in the literature regarding their bonding and reactivity [1]. Since allenes in rings with less than nine members can not adopt their preferred linear geometry, several structures have been proposed for singlet 1,2-cyclohexadiene, suggesting that they could exist as dipolar or diradical species [2].
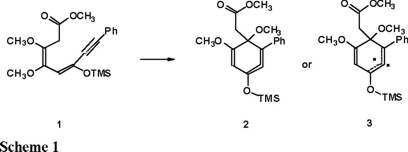
We have recently reported [3] using Hartree-Fock Self Consistent Field (HF) and single reference frozen-core second order Möller-Plesset (MP2) ab initio calculations that the ring closure of dienyne 1 generates an intermediate that can best be described as the cyclic allene 2 and not the diradical form 3 (Scheme 1), in agreement with earlier literature reports in which the cyclic allene 1,2-cyclohexadiene is favored over other species [4].
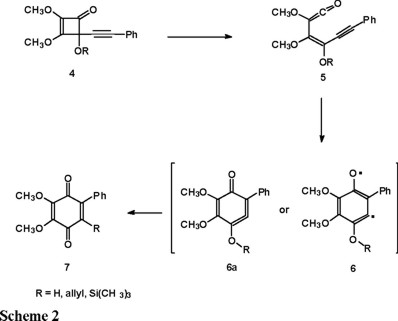
In the light of the above result, we decided to analyze the last step of the original Moore's benzoquinone synthesis [5] (Scheme 2) using ab initio calculations. The main goal of this study is to characterize the nature of the intermediate obtained by the ring closure of 5 as the diradical 6, as it has been postulated in the past [6], or the cyclic allene 6a, in analogy with our previous results. We present results concerning the lowest singlet and triplet states since theses multiplicities characterize the cyclic allene and the diradical species respectively.
Theoretical calculations
In order to achieve our goal, we performed HF, UHF and MP2 full geometry optimizations using the standard 6-31G** basis of the Gaussian-94 program [7] on both structures 6 and, 6a presented in Scheme 2 and on simpler models of these in which the phenyl group is replaced by a hydrogen atom respectively and referred to as structures 8 and 9. The triplet species were also calculated for the simpler models using unrestricted Hartree-Fock (UHF) and unrestricted Möller-Plesset (UMP2) calculations.
It must be said that an open-shell singlet state (the counterpart of the lowest triplet state) must also exist and that we have tried to obtain this state through UHF calculations starting from the triplet state; however even though during the first few SCF cycles the open-shell state was lower in energy, after 8 iterations the closed-shell singlet state turned out to be favored. The stability of this closed-shell solution was tested (including HF instabilities) by using the Stable=Opt option in the G98 program.
The most important result of these calculations is that the strained cyclic allene structure 6a corresponds to the ground state of the intermediate rather than the diradical 6. For structure 8 our UHF calculations predict a triplet ground state which lies 27.15 kcal / mol below the optimized singlet structure, while at the UMP2 level the singlet is favored as the ground state by 19.6 kcal / mol. These findings are in agreement with the common observation that the HF description favors the higher multiplicities but electron correlation effects are larger for the closed-shell species and thus we expect that higher level calculations will not modify the present results. It is important to mention that for structure 9 we did not anticipate a low energy triplet state and in fact, our calculations predict a singlet ground state 18.9 kcal / mol below the triplet at the UHF level and 81.0 kcal / mol at the UMP2 level. It is interesting to know that correlation effects are crucial to obtain reliable singlet-triplet energy gaps for these molecules.
We are confident that the conclusions derived for the simpler models 8 and 9 should hold for the actually synthesized structure 7 derived from intermediate 6 or 6a since we have performed HF and MP2 optimizations on the singlet states of these latter molecules and have found that the presence of the phenyl group has a very small effect on the geometry (i.e. the internuclear distances between carbon atoms that define the cyclic allene structure). This implies that in Scheme 1 the reaction intermediate is a singlet cyclic allene and there is little chance that the triplet diradical can play a role in the reactivity of these systems. This is contrary to the usual assumptions made for these type of chemical reactions. The existence of diradical character has been found for structure 8 at the UHF level for the triplet state by graphically analyzing the spin density and it corresponds to the chemical drawing depicted for structure 6 (without the phenyl group) in agreement with chemist's conventional wisdom.
Structures
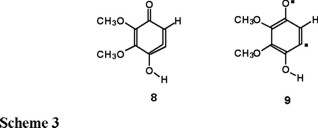
The analysis of the optimized geometries for structures 8 and 9 indicates significant differences depending on the level of theory used and on the multiplicity of the state considered. First we discuss the distinctive features of the allene moiety in the singlet state using the molecular structures 8 and 9 of Scheme 3. We must stress that the ground state singlet 8 turned out to be —as it was confirmed by the previously mentioned CASSCF calculations- the closed-shell cyclic allene and that the diradical species 9 lie well above the cyclic allene.
The conventional picture would propose three double bonds linking atoms C2-C3, C3-C4 and C5-C6 and a single bond between C4-C5 in the ring. However at the HF level structure 9 is better describes as structure 10 where the bond lengths C3-C4 and C4-C5 are intermediate between a single and a double bond (1.43 Å). The previously mentioned conventional picture is reproduced at the MP2 level as can be seen in structure 8 of Scheme 4. At both levels of theory we find that the six-membered ring is non-planar, it has a conformation which can be described as 'boat' with carbon atoms C4 and C1 out of the plane, the former being significantly more bent.
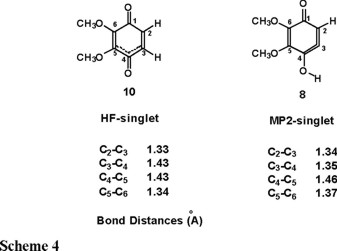
The final feature which we consider worth of mentioning is the different orientation of the methoxy groups depending on the level of calculation. At the HF level the methoxy group linked to carbon C6 lies in the plane of the ring while the methoxy linked to carbon C5 is facing downwards. Even though we used this geometry as starting point for the MP2 optimization, the converged structure has the methoxy linked to C5 planar and the other methoxy facing upwards. It should be mentioned here that we did not search for all possible stable conformers for these molecules.
The most striking features for the triplet state of structure 10 (Scheme 3) are that the six-membered ring becomes planar and the C3-C4 and C4-C5 bond lengths (Scheme 5) change dramatically compared with the singlet state.
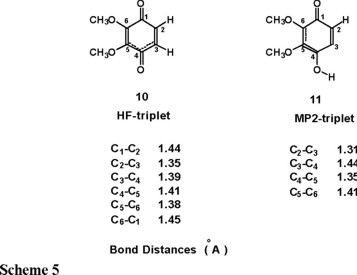
At the HF level, structure 10, the C3-C4 distance is reduced to a value intermediate between a single and a double bond and a similar change is observed in the C4-C5 bond but to a lesser extent. At the MP2 level, structure 11, the single or double bond nature of the C3-C4 and C4-C5 bonds is the opposite of what was found for the singlet state, i.e., the C3-C4 bond is weakened while the C4-C5 bond becomes a double bond. Comparing the quantitative geometrical data of the structures in Scheme 4 and Scheme 5, we must conclude that the HF description leads to completely unreliable results for the cyclic allene (singlet) as well as for the diradical (triplet) species. Thus electronic correlation effects are crucial in describing these molecular systems
This type of molecules represent a true challenge for modern quantum chemistry given the competition between closed and open-shell structures (singlet as well as triplet states) and their size and stereochemical complexity.
We are presently working on systematic description of these molecules using a variety of ab initio and DFT methods in analogy to the work which is currently being done on the Myers-Saito and Schmittel cyclizations [8], cyclizations which have also been the subject of several important recent theoretical studies. However, one of the important differences with our problem at hand is that the active electrons in these structures are always found on carbon atoms, while in our intermediate molecule an oxygen atom linked to a six-membered ring is involved.
In the newest and most complete study dealing with the highly reactive nature of the diradicals involved in the Myers-Saito and Schmittel cyclizations the emphasis is therefore put only on the study of singlet and triplet open-shell species [8c]. However, if the main question is aimed at finding the most stable structure of these type of molecules, we think that crucial information is missing regarding the energetic position of the closed-shell singlet structure, which was not envisaged in that work. In fact, as was shown in a pioneering ab initio work concerning the 1,2, cyclohexadiene by Schmidt et al. [9], it is the closed-shell singlet structure the one which is favored over the singlet open-shell diradical species. However we must stress that the latter one can actually play a major role in the reactivity of this molecule.
In order to give further support to our results and conclusions, we have performed state-averaged CASSCF calculations on the two lowest roots for the singlet species, we found that the lowest root corresponds to the closed -shell species, as we had already shown. However, in order to obtain a more reliable energy difference between the closed-shell and the singlet diradical, we tried to optimize the orbitals for the second root aiming at state specific CASSCF calculation for the singlet diradical (using the triplet biradical orbitals as starting point). Unfortunately this was not possible as convergence problems were found. Pursuing these calculations further would be a very time consuming task and is out of the scope of this work.
Conclusions
Our calculations show that the most stable intermediate in the Moore benzoquinone synthesis is the cyclic allene 6a and not the diradical 6. We have confirmed the presence of the diradical as a triplet at 19.6 Kcal / mol above the cyclic allene. We have also observed very strong changes on the electronic structure as the molecule passes from singlet to triplet nature, this being based on the fact that the lowest singlet is not related to the two-open-shell diradical. Finally, there are also strong effects (structurally and energetically) arising from the non-dynamical electronic correlation on both, the allene and the diradical which show the limitation of using single-configuration based methods on these systems.
Acknowledgements
The authors wish to thank CONACyT for providing financial support for this work through projects 3112P-E9607, 34673-E and 3446E.
References
1. Johnson, R.P. in "Molecular Structures and Energetics", Vol 3, J.F. Liebman and A. Greenberg, Ed., Verlag Chemie International, 1985. [ Links ]
2. (a) Moore, W.R.; Moser, W.R. J. Am. Chem. Soc. 1970, 92, 5469-5474. [ Links ] (b) Dillon, P.W.; Underwood, G.R. J. Am. Chem. Soc. 1974, 92, 779-791. [ Links ] (c) Schmidt, M.W.; Angus, R.O.; Johnson, R.P. J. Am. Chem. Soc. 1982, 104, 6838-6839. [ Links ]
3. Fernández-Zertuche, M.; Hernández-Lamoneda, R.; Ramírez-Solís, A. J. Org. Chem. 2000, 65, 5207-5211. [ Links ]
4. Bottini, A.T.; Corson, F.P.; Fitzgerald, R.; Frost, K.A.,II Tetrahedron 1972, 28, 4883-4904. [ Links ]
5. a) Moore, H.W.; Xiang, Y. J. Org. Chem. 1996, 61, 9168-9177. [ Links ] (b) Xiang, Y.; Xia, H.; Moore, H.W. J. Org. Chem. 1995, 60, 6460-6467. [ Links ] (c) Chow, K.; Nguyen, N.V.; Moore, H.W. J. Org. Chem. 1990, 55, 3876-3880. [ Links ] (d) Chow, K.; Moore, H.W. ibid. 1990, 55, 370-372.
6. (a) Moore, H.W.; Yerxa, B.R. Adv. Strain. Org. Chem. 1995, 4, 81-162. [ Links ] (b) Foland, L.D.; Karlsson, J.O.; Perri, S.T.; Schwabe, R.; Xu, S.L.; Patil, S.; Moore, H.W. J. Am. Chem. Soc. 1989, 111, 975-989. [ Links ] (c) Karlsson, J.O.; Nguyen, N.V.; Foland, L.D.; Moore, H.W. ibid 1985, 107, 3392-3393.
7. Gaussian 94, Revision C.3, M. Frisch, J.; Trucks, G.W.; Schlegel, H.B.; Gill, P.M.W.; Johnson, B.G.; Robb, M.A.; Cheeseman, J.R.; Keith, T.; Petersson, G.A.; Montgomery, J.A.; Raghavachari, K.; Al-Laham, M. A.; Zakrzewski, V. G.; Ortíz, J.V.; Foresman, J. B.; Cioslowski, J.; Stefanov, B. B.; Nanayakkara , A.; Challacombe, M.; Peng, C.Y.; Ayala, P.Y.; Chen, W.; Wong, M.W.; Andres, J.L.; Replogle, E. S.; Gomperts, R.; Martin, R.L.; Fox, D.J.; Binkley, J. S.; Defrees, D. J.; Baker, J.; Stewart, J. P.; Head-Gordon, M.; González, C.; Pople, J. A. Gaussian, Inc., Pittsburgh PA, 1995. [ Links ]
8. (a) Engels, B.; Hanrath, M. J. Am. Chem. Soc. 1998, 120, 6356-6361. [ Links ] (b) Engels, B.; Lennartz, C.; Hanrath, M.; Schmittel, M.; Strittmatter, M. Angew. Chem. Int. Ed. 1998, 37, 1960-1963. [ Links ] (c) Schreiner, P.R., Prall, M. J. Am. Chem. Soc. 1999, 121, 8615-8426, [ Links ] and references therein.

