










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Sociedad Química de México
versión impresa ISSN 0583-7693
Rev. Soc. Quím. Méx vol.46 no.1 Ciudad de México ene./mar. 2002
Investigación
Improved Synthesis of (20S)-3β-Acetoxy-23,24-dinorchol-5-en-22-aldehyde
Cirilo García,* Josefina Tapia and Humberto Cervantes
Universidad Autónoma Metropolitana, Departamento de Ciencias Básicas, Área de Química, Av. San Pablo #180, Col. Reynosa Tamaulipas México 02200 D.F. Tel. 5318-9497; Fax. 5318-9540. E-mail: gmc@correo.azc.uam.mx
Recibido: 22 de enero del 2002.
Aceptado: 27 de marzo del 2002.
Abstract
Commercial 3β-acetoxy-23,24-dinorchol-5-en-22-oic acid was transformed into (20S)-3β-acetoxy-23,24-dinorchol-5-en-22-aldehyde in a two-step process. The key step was reduction of the imidazolide derived from the above mentioned acid, which invariably produced the target aldehyde and 22-hydroxy-23,24-dinorchol-5-en-3β-yl acetate. In adittion to these products, the work-up of the organic material from the latter reaction, afforded pregn-5-en-3β-yl acetate along with the mixture of the former acid and its unreacted imidazolide. Oxidation of 22-hydroxy-23,24-dinorchol-5-en-3β-yl acetate with pyridinium chlorochromate and recycling of the mixture acid/imidazolide, enhances the practical value of this process. Structural analysis, based on 1H NMR spectroscopy and polarimetry of intermediate and final products, and observations on the stability of the imidazolyl derivative and aldehyde are briefly discussed.
Keywords: Pregn-5-ene-20-carboxaldehyde; 3-acetoxy-bisnor-5-cholenaldehyde, dinorcholenic acid, bisnorcholenic acid, steroidal imidazolid.
Resumen
El presente estudio describe la transformación del ácido 3β-acetoxi-23,24-dinorcol-5-en-22-oico al (20S)-3β-acetoxi-23,24-dinorcol-5-en-22-aldehido. La transformación se realizó a través de la (20S)-22-(1H-imidazol-1-il)-3β-acetoxi-23,24-dinorcol-5-en-22-ona, cuya reducción con hidruro de tri-t-butoxialuminio-litio dio el aldehido mencionado arriba y el acetato de 22-hidroxi-23,24-dinorcol-5-en-3β-ilo, el imidazólido sin reaccionar y el ácido esteroidal de partida. La posibilidad de reciclar el ácido esteroidal y el imidazólido, y de oxidar el acetato de 22-hidroxi-23,24-dinorcol-5-en-3β-ilo al producto objetivo, son las características distintivas del presente método. Se incluye una breve discusión del análisis estructural de los productos intermediarios y finales, y acerca de la estabilidad del imidazólido y del aldehido deseado.
Palabras clave: Pregn-5-en-20-carboxaldehido, 3-acetoxi-bisnor-5-colenaldehido, ácido dinorcolénico, ácido bisnorcholénico, imidazólido esteroidal.
Introduction
A useful starting material for the synthesis of biologically active steroids is (20S)-3β-acetoxy-23,24-dinorchol-5-en-22-aldehyde (1). The carbonylic carbon of 1 allows to work on the side chain, while the protected 3-hydroxy group and C5-C6 double bond allow modifications on rings A and B, respectively. Aldehyde 1 is not commercially available but it can be prepared either by ozonolysis of stigmasterol derivatives [1,2] or by reduction of 23,24-dinorcholenic acid derivatives [3-5]. As part of our current work on stereocontrolled condensations, it has been necessary to search the most suitable procedure to prepare several grams of aldehyde 1. Experimental results showed that reduction and oxidation of compounds derived from 3β-acetoxy-23,24-dinorcho-5-en-22-oic acid (2) afford better results than ozonolysis of stigmasterol derivatives; therefore, the present contribution is focused on the reinvestigation of the procedure published by McMorris in 1970 [3]. Careful observations on stability, optical relation and 1H NMR data of intermediate and final products are discussed.
Results and discussion
The transformation of commercially available 2 into aldehyde 1, was carried out as it is outlined in Scheme 1: acid 2 was treated with 1,1'-carbonyldiimidazole at 60 °C to give imidazolide 3. This reaction was performed several times with different ratios of acid 2 to 1,1'-carbonyldiimidazole, and the corresponding results are shown in Table 1. Imidazolide 3, was isolated as colorless small needles and its structure and purity were corroborated by 1H NMR spectroscopy. Imidazolide 3 was reduced with lithium tri-tert-butoxyaluminohydride at 25 °C. All runs were performed with 10 % excess of the reducing agent. After work-up and flash chromatography of the crude product, aldehyde 1 was isolated in 67.4 % yield based on initial imidazolide 3. In addition to 1, four more components were isolated and characterized as alcohol 4 (15.3 %), pregnene 5 (0.3 %), imidazolide 3 (1.8%) and acid 2 (11.2%), respectively. Alcohol 4 was transformed into aldehyde 1 with pyridinium chlorochromate [4] in 86 % yield. Specific rotation, melting point and 1H nmr spectra of aldehyde 1 prepared either by reduction of 3 or by oxidation of 4 were similar, indicating that both samples had the same configuration at C-20.

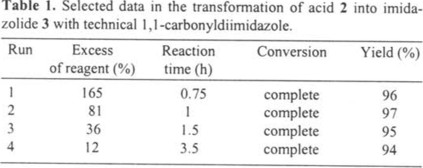
Specific rotation for aldehyde 1 having 3β-OTHP, 3β-TBDMS or 3β-OAc groups has been reported elsewhere; but in some cases, authors have not mentioned the concentration [6] or they have overlooked typographic errors on their data [5]. Then, we decided to include the specific rotation in the present contribution. The data are presented in Table 2 using standard units [7].
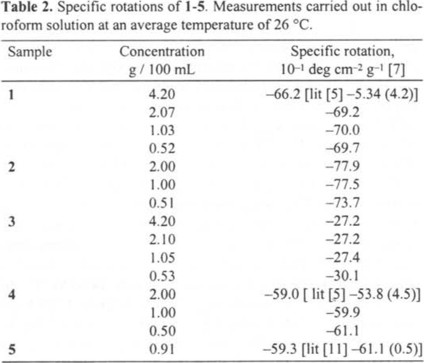
Preparation of imidazolide 3. Staab and Bräunling [8] published that equimolar ratio of low molecular weight caboxylic acids and pure 1,1'-carbonyldiimidazole give high yields of imidazolides. In our case 12 % excess of technical carbonyldiimidazole, which contains about 90 % weight of 1,1-carbonyldiimidazole [9], was enough to afford quantitative transformation of acid 2 into imidazolide 3 within 3.5 h. Therefore, the two-fold excess of reagent used by McMorris [3] is unnecessary. Isolation of 3 was essentially done as described before [3], but we found more convenient to dissolve and recrystalize the wet crude product from CH2Cl2 instead to dry it over P2O5. This modification affords pure imidazolide 3 in a short time.
Preparation of aldehyde 1. Reaction of 3 with tri-tert-butoxialuminohydride was essentially carried out as described by McMorris [3]. Aldehyde 1 was isolated as the main product, its physical properties and structure are in agreement with the shown configuration. This means that reduction occurs with retention of configuration and that silica gel does not catalyze epimerization of carbon C-20 [10]. Crystallized aldehyde 1 prepared by this method can be stored at 0 °C under nitrogen for several weeks without decomposition; however, it undergoes gently oxidation by air. Indeed, when a solution of pure 1 was stirred in an open container during 15 days, acid 2 was isolated in 24 % yield along with 61 % of unreacted 1.
As expected, reductive action of tri-tert-butoxialuminohydride transforms 1 into alcohol 4. Despite the low excess of tri-tert-butoxialuminohydride, we used 10 % instead of reported 56 % [3] alcohol 4 was invariably isolated in 15.3 % yield in all runs performed between 5 and 25 °C. The high cost of these steroids, and to increase the global yield of 1. After this transformation, the yield of aldehyde 1 based on the initial amount of imidazlide 3, results higher than that reported by McMorris [3], 80 versus 75 %. Evidently, the present result is superior because recovered acid 2 and unreacted imidazolide 3 can be recycled.
Isolation of acid 2 from the reductive process of 3, made us to search its plausible formation route. Our preliminary results, shows that hydrolysis of imidazolide 3 occurs faster than aerial oxidation of aldehyde 1; therefore, we propose that acid 2 is formed during the work-up and purification process of 1. A complete study on the hydrolysis of imidazolides will be the subject of future contribution.
Formation of pregnene 5 as a by-product in the reduction of 3 with tri-tert-butoxialuminohydride has not been reported before. Despite its low yield, pregnene 5 was isolated in pure form, its melting point specific rotation and 1H nmr spectrum are identical with reported data [11, 12]. Our observations on the stability of aldehyde 1 and acid 2 under normal laboratory conditions, suggest that 5 could come from decarbonylation of aldehyde 1 in the presence of tri-tert-butoxialuminohydride; however, further experimental work is necessary to explain this transformation. A closely related study, is the radical induced reduction of steroidal phenylcarboselenoates and phenyl selenocarbonates with tributyltin hidride [12].
1H NMR. The 1H NMR spectrum of acid 2 consists of a broad signal centered at 9.630 ppm assigned to hydroxylic proton a doublet at 5.374 ppm (J = 4.5 Hz) assigned to H-6, a multiplet centered in 4.602 ppm attributed to Hα-3, a doublet of quartets in 2.434 ppm (Jq = 6.8 Hz and Jd = 10.5 Hz) assigned to Hβ-20, three single signals at 2.036, 1.022 and 0.707 ppm, assigned to protons of acetyl, methyl C-19 and methyl C-18, respectively, and a doublet at 1.244 ppm (J = 6.8 Hz) assigned to protons of methyl C-21.
The transformation of acid 2 into imidazolide 3 can be easily recognized by inspection of their 1H NMR spectra. The imidazolyl moiety gives an ABX system [13] and induces downfield shifts in the resonance signals of the nearest protons. The magnitude of this deshielding is nearly proportional to the distance between such protons and imidazolyl group, as can be observed when the chemical shifts of Hβ-20, methyl C-21 and C-18 on the spectrum of 2 are subtracted from those on the spectrum of 3: Δδ values are 0.613, 0.127 and 0.087 ppm, respectively. As expected, the effect of imidazolyl on the resonance signals of H-6, Hα-3, methyl C-19 and acetyl group is almost negligible.
1H NMR spectrum of aldehyde 1, shows a doublet at 9.572 ppm (J = 3.3 Hz) assigned to H-22, a multiple centered at 2.362 ppm for Hβ-20 (partially overlapped with the signal attributed to HαHβ-4 [14]) and a doublet at 1.138 ppm (J = 6.8 Hz) for methyl C-21. It must be noted that signals due to Hβ-20 and methyl C-21 are shielded compared with the corresponding signals of imidazolide 3 and acid 2. The 1H NMR spectra of 1 obtained either by reduction of 3 or by oxidation of alcohol 4, gave only the downfield signal characteristic of aldehydic proton and therefore, we concluded that we obtained a single diastereomer [10]. Resonance signals of H-6, Hα-3 and methyl C-18, C-19 and acetyl groups undergo very small changes.
The spectrum of alcohol 4 shows the AB pattern of the expected ABX system, due to methylenic protons C-22 and methine C-20, (δA 3.640, δB 3.361, JAB = 10.5 Hz, JAX = 3.2 Hz, JBX = 7.0 Hz) and a doublet at 1.052 ppm (J = 6.6 Hz) assigned to methyl C-21. Resonance signals of H-6, Hα-3, acetyl, methyl C-18 and C-19, show almost the chemical shifts mentioned above for acid 2.
The spectrum of pregnene 5 shows the signals for H-6, Hα-3, acetyl, and methyl C-19 with the multiplicity and almost the same chemical shifts described above for acid 2. The main feature on this spectrum is the triplet at 0.875 (J = 7.2 Hz) which is assigned to methyl C-21 and the single signal at 0.579 ppm assigned to methyl C-18. These assignments are in agreement with reported data for authentic pregnene 5 [11, 12]; and furthermore, we verified that the 13C NMR spectrum of 5 contains the 23 expected signals.
Conclusions
We have improved the two-step preparation of 22,23-dinorchol-5-en-22-aldehyde 1 reported by McMorris [3] and have measured the specific rotation, infrared and 1H NMR spectra of the intermediate and final products. Aldehyde 1 is readily oxidized by air and presumably deoxigenated by exposure to tri-tert-butoxyaluminohydride and light.
Experimental
General. Reactions were performed under nitrogen atmosphere using dry THF evaporated from sodium and benzophenone. Solvents used for extraction and column chromatography were freshly distilled. Technical grade 1,1'-diimidazol carbonyl, 90 % [9] and tri-tert-butoxialuminohydride (0.5 M solution in THF), were purchased from Aldrich while 3β-acetoxy-23,24-dinorchol-5-en-22-oic acid was purchased from Steraloids. These chemicals were used without further purification. For tlc and the so called flash chromatography [15] were used Merck sheets of Alufolien and silica gel 230-400 mesh, respectively.
Specific rotation was measured in chloroform solutions in a PerkinElmer 341 polarimeter using a 6 mL cell, 10 mm long. Solutions were prepared in 10 mL volumetric flasks. The lectures were taken at a mean temperature of 26 °C and units are 10-1 deg cm-2 g-1 [7]. Infrared spectra were recorded on a FT Vector 33 equipped with horizontal ATR accessory having a 45° ZnSe crystal. 1H NMR spectra were recorded on a Bruker DPX 300 MHz spectrometer equipped with a 5 mm 1H probe. A weighed amount of the substrate was dissolved in CDCl3 to give a 0.05-0.1 M solution at 25 °C. The chemical shifts were referenced to internal TMS. Typical 1H NMR spectra were obtained using pulse width of 11.5 µs (45°), a pulse delay of 1 s, 32 transients in a spectral width of 3592 Hz digitized into 64K data points, resulting in a digital resolution of 0.11 Hz per point. NMR data is given in standard format: δ units, signal multiplicity, coupling constants in Hz and assignment.
Preparation of (20S)-22-(1H-imidazol-1-yl)-3β-acetoxy-23,24-dinorchol-5-en-22-one (3). In a three-neck round-bottom flask of 500 mL, equipped with condenser, addition funnel and a stirring bar, were added 1,1'-carbonyldiimidazole (5.194 g, 28.829 mmol) and free-water THF (60 mL). A solution of 3β-acetoxy-23,24-dinorchol-5-en-22-oic acid (2) in dry THF (10 g, 25.740 mmol / 50 mL) were transferred into the addition funnel by a double-ended needle. This solution was added with stirring to the 1,1'-carbonyldiimidazole in 10 min at 25 °C. A further 15 mL portion of THF was added through the addition funne, and the resulting mixture was heated under reflux for 3.5 h. Then, the solvent was evaporated under vacuum and the resulting solid was poured into chilled water (250 mL), stirred for 5 min and filtered through a Buchner funnel with glass fritted disc. After a second washing with 200 mL water, the white solid was dissolved in CH2Cl2 (100 mL) and the resulting emulsion was dried (Na2SO4) and concentrated in rotavapor up to a tenth of the initial volume and left on standing at 25 °C. Crystals of imidazolide 3, small colorless needles (m.p. 223-224 °C, lit. [3] 220-223 °C), were filtered by suction and mother liquors were subjected to further crystallization. The total amount of dry imidazolide 3 was 10.610 g. Its specific rotation is given in Table 2. IR (neat) νmax 1717.8, 1244.4 and 1198.7 cm-1. 1H NMR, δ 8.197 (1H, br s, H-2 imidazolyl); 7.509 (1H, t, J = 1.4 Hz, H-5 imidazolyl); 7.115 (1H, br s, H-4 imidazolyl); 5.377 (1H, d, J = 5.1 Hz, H-5); 4.6 (1H, m, Hα-3); 3.047 (1H, dq, J = 6.9 and 9.7 Hz, Hβ-20); 2.038 (3H, s, Ac); 1.371 (3H, d, J = 6.8 Hz, Me C-21); 1.038 (3H, s, Me C-19) and 0.796 (3H, s, Me C-18).
Preparation of (20S)-3β-acetoxy-23,24-dinorchol-5-en-22-aldehyde (1). Method A. In a three neck round bottom flask of 500 mL equipped with addition funnel and a stirring bar, were placed the imidazolide 3 (11.8 g, 26.9 mmol) and dry THF (120 mL). This mixture formed a white emulsion which was chilled to 5 °C. In the addition funnel was placed dry THF (40 mL) and the required amount of tri-tert-butoxyaluminohydride (29.6 mL, 1 M in THF) and then it was slowly added to the suspension of 3, in 70 min with stirring. Further 10 mL of THF were added through the addition funnel and the temperature was raised to 25 °C. This mixture was stirred at 25 °C until all the solid disappeared and then for further 30 min at the same temperature. The solvent was evaporated under vacuum and the yellowish residue was mixed with chilled 1 N HCl solution (250 mL). The resulting suspension was filtered by suction and the white solid was washed with cold water (120 mL). The wet solid was poured with stirring into CH2Cl2 (200 mL), the organic solution was dried (Na2SO4) and concentrated in rotavapor. Tlc of CH2Cl2 solution consisted of five spots having the following Rf values: 0.607, 0.428, 0.167, 0.131 and 0.05 (hexanes-AcOEt, 4:1). A rough separation of the two less polar components was carried out by column chromatography (silica gel, CH2Cl2) and further elution with AcOEt afforded the mixture of the three more polar components. The main component, aldehyde 1 found in CH2Cl2 fractions, was subjected to successive purification by column chromatography (hexanes-AcOEt 46:4) to afford 6.755 g of small colorless leaflets (m.p. 114-116 °C, hexanes-AcOEt, lit. [13] 113-116 °C). IR (neat) νmax 1725.3 1255.1 and 1039.5 cm-1. 1H NMR of 1, δ 9.572 (1H, d, J = 3.3 Hz, H-22); 5.376 (1H, br d, J = 5.0 Hz, H-6); 2.362 (1H, ddq, J = 6.8 Hz, J = 3.3 Hz, J = 10.2 Hz, Hβ-20); 2.036 (3H, s, Ac); 1.138 (3H, d, J = 6.8 Hz, methyl C-21); 1.028 (3H, s, methyl C-19) and 0.731 (3H, s, methy C-18). The less polar component, characterized as pregn-5-en-3β-yl acetate (5) was also purified by column chromatography (hexanes-AcOEt 48:2) and recrystallized from methanol to obtain 28 mg of small colorless needles (m.p. 144-147 °C, lit. [11] 148-149 °C). IR (neat) νmax 1729.4, 1239.5 and 1028.3 cm-1. 1H NMR of 5, δ 5.383 (1H, br d, J = 4.9 Hz, H-6); 4.606 (1H, m, Hα-3); 2.035 (3H, s, methyl Ac); 1.026 (3H, s, methyl C-19); 0.875 (3H, t, J = 7.2 Hz, methyl C-21) and 0.579 (3H, s, methyl C-18). The component that produces the spot of Rf 0.167 (see above), identified as 22-hydroxy-23,24-dinorchol-5-en-3β-yl acetate (4), was also purified by column chromatography (CH2Cl2) and recrystallized from ethanol to afford 1.546 g (small colorless needles, m.p. 149-151 °C , lit. [5] 153-154 °C). IR (neat) νmax 3271.0, 1726.8, 1259.4 and 1041.4 cm-1. 1H NMR of 4, δ 5.375 (1H, br d, J = 4.2 Hz, H-6); 4.602 (1H, m, Hα-3); 3.640 (1H, dd, JAB = 10.5 Hz, JAX = 3.2 Hz, HA-22); 3.361 (1H, dd, JAB = 10.5 JBX = 7.0, HB-22); 2.035 (3H, s, methyl Ac); 1.052 (3H, d, J = 6.6 Hz, methyl C-21); 1.022 (3H, s, methyl C-19) and 0.703 (3H, s, methyl C-18). The mixture of the two more polar components was obtained as a yellowish solid. 1H NMR analysis of this mixture showed to be composed by 86 % mol of acid 2 and 12 % mol of imidazolide 3, and therefore, it was hydrolyzed and the resulting solid recrystallized from CH2Cl2 to afford 1.364 g of acid 2 (colorless leaflets, m.p. 237-241 °C).
Method B. In a 100 mL round bottom flask with a stirring bar, was placed a weighed amount of pyridinium chlorochromate (0.972 g, 4.5 mmol) and covered with dry CH2Cl2 (15 mL). Then, a solution of alcohol 4 (1.12 g, 3 mmol in 30 mL CH2Cl2) was added in one portion at 25 °C, and the resulting dark mixture was stirred for 1.5 h. After this time, tlc showed a single spot with Rf 0.5 (hexanes-AcOEt 85:15). The dark mixture was passed through a short column packed with silica gel 70-230 mesh. Elution with CH2Cl2 followed by evaporation of the solvent and recrystallization of the yellowish solid (hexane-CH2Cl2), afforded 0.960 g of a white powder whose m.p., specific rotation, IR and 1H NMR spectra resulted identical with those of aldehyde 1 prepared by method A.
Acknowledgments
The authors thank the Consejo Nacional de Ciencia y Tecnología de México for financial support (proyecto 32261-E).
References
1. Centolella, A. P.; Heyl, F. W.; Herr, M. E. J. Am. Chem. Soc. 1948, 70, 2953-2954. [ Links ]
2. Fryberg, M.; Oehlschlager, A. C.; Unrau, A. M. Tetrahedron 1971, 27, 1261-1274. [ Links ]
3. McMorris, T. C. J. Org. Chem. 1970, 35, 458-460. [ Links ]
4. Coldwell, R. D.; Trafford, D. J. H.; Varley, M. J.; Kirk, D. N.; Makin, H. L. J. Steroids 1990, 55, 418-432. [ Links ]
5. Ishiguro, M.; Saito, H.; Sakamoto, A.; Ikekawa, N. Chem. Pharm. Bull. 1978, 26, 3715-3721. [ Links ]
6. Edwards, J. A.; Mills, J. S.; Sundeen, J.; Fried, J. H. J. Am. Chem. Soc. 1969 91, 1248-1249. [ Links ]
7. Eliel E. L.; Wilen, S. H. in: Stereochemistry of organic compounds, John Wiley & Sons, Inc., N.Y., pp. 6-8, 1994. [ Links ]
8. Staab, H. A.; Bräunling. H. Justus Liebigs Ann. Chem. 1962, 654, 119-130. [ Links ]
9. Swessel, D. Aldrich. Private communication, 2001. [ Links ]
10. For epimerization of aldehyde 1 in aluminium oxide, see Sato, Y.; Sonoda, Y.; Saito, H. Chem. Pharm. Bull. 1980, 28, 1150-1156. [ Links ]
11. Clive, D. L. J.; Chittattu, G. J.; Farina, V.; Kiel, W. A.; Menchen, S. M.; Russell, C. G.; Singh, A.; Wong, C. K; Curtis, N. J. J. Am. Chem. Soc. 1980, 102, 4438-4447. [ Links ]
12. Pfenninger, J.; Heuberger, C.; Graf, W. Helv. Chim. Acta 1980, 63, 2328-2337. [ Links ]
13. Integrated Spectral Data Base for Organic Compounds Page. http://www.aist.go.jp/RIODB/SDBS/menu-e.html (SDBS No. 399, accessed Dec 2001).
14. Dawe, R. D.; Wright J. L. C. Can. J. Chem. 1987, 65, 666-669. [ Links ]
15. Still, W. C.; Kahn, M.; Mitra, A. J. Org. Chem. 1978, 43, 2923-2925. [ Links ]

