










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista mexicana de cardiología
versión impresa ISSN 0188-2198
Rev. Mex. Cardiol vol.24 no.2 México abr./jun. 2013
TRABAJO DE REVISIÓN
Tetralogía de Fallot. Actualización del diagnóstico y tratamiento
Tetralogy of Fallot. Diagnosis and treatment update
Carlos Alva Espinosa*
* Cardiólogo y cardiólogo pediatra. Director de Planeación Enseñanza e Investigación del Hospital Regional de Alta Especialidad, Secretaría de Salud, Ixtapaluca, Estado de México.
Dirección para correspondencia:
Dr. Carlos Alva Espinosa
Carretera México Puebla km 34.5 Ixtapaluca,
Estado de México.
Tel: 5972-9800 Ext: 1202 Cel: 55 2129-4497
E-mail: carlosalvaespinosa@yahoo.com.mx
RESUMEN
Casi siete décadas han transcurrido desde la primera paliación quirúrgica a una enferma con tetralogía de Fallot (TOF). Esta es la cardiopatía compleja más tratada y mejor conocida. El tiempo ha permitido ver los resultados a largo plazo de los diferentes abordajes quirúrgicos; estas observaciones han permitido a los equipos médico-quirúrgicos hacer modificaciones, en especial la corrección primaria inicial entre los 6 y 12 meses de vida versus en dos tiempos. El desarrollo tecnológico en las herramientas diagnósticas, operatorias y posoperatorias también ha mejorado los resultados. En la actualidad es posible esperar una sobrevivencia del 90% de los niños con TOF a 30 años después de la corrección quirúrgica, con una calidad de vida normal en muchos de ellos; sin embargo, la necesidad de reoperación y las complicaciones tardías incluyendo las arritmias y la muerte súbita son problemas no resueltos. Este artículo es una revisión sobre la tetralogía de Fallot que incluye los aspectos históricos, diagnósticos, la evolución de los criterios quirúrgicos, las controversias actuales sobre los mismos y sus resultados, el papel de la cardiología intervencionista, el desfibrilador implantable y cómo se vislumbra el futuro próximo.
Palabras clave: Tetralogía de Fallot, México, historia, diagnóstico, abordaje quirúrgico.
ABSTRACT
Tetralogy of Fallot is the best known and most treated complex congenital heart disorder. The first palliative surgery in a patient with tetralogy of Fallot was performed almost seven decades ago. In this time period specialists have established the long-term results of different surgical techniques and allowed the medical and surgical teams to introduce modifications, particularly the complete correction performed between 6 and 12 months of life versus the two-stage approach. The technological advances in diagnostic, surgical and postsurgical tools have also improved the results. Today 90% of children diagnosed with tetralogy of Fallot are expected to survive to 30 after the surgical correction, many of them leading a normal life. However, the requirement for further surgical interventions, late cardiac complications, including arrhythmias, and sudden death are unsolved problems. This article reviews the historic aspects and the evolution of the surgical criteria in tetralogy of Fallot, current results and controversies, the implantable defibrillator, the role of interventional cardiology and future directions.
Key words: Tetralogy of Fallot, Mexico, history, diagnosis, surgical approach.
Introducción
Han transcurrido 69 años desde que Eileen Saxon, una niña de quince meses de edad, entró a quirófano para recibir la primera fístula sistémico-pulmonar que salvó su vida. Eileen tenía tetralogía de Fallot (TOF) y fue la primera enferma con una cardiopatía congénita cianógena tratada quirúrgicamente en 1944.1 Fue una idea de Helen Taussig, narrada por ella misma2 y surgida de dos observaciones; primero, notó que los bebés que nacían con TOF y soplo continuo de conducto tenían menos cianosis hasta que éste se cerraba y la segunda, a través de la fluroscopia (la mejor herramienta en aquellos tiempos) observó flujo pulmonar disminuido en los pulmones de estos enfermos. Bajo este razonamiento, Helen recomendó a Blalock la conveniencia de crear un conducto arterioso en estos niños. Alfred Blalock, convencido, realizó la primera fístula, con la ayuda del brillante técnico en cirugía Vivien Thomas en el Johns Hopkins de Baltimore.
Durante el transcurso de las siete décadas siguientes, una gran experiencia y una vasta información se han acumulado en el mundo. El propósito de este artículo es revisar y puntualizar los aspectos más relevantes de esta fascinante entidad, emblemática de las cardiopatías congénitas en el diagnóstico, las indicaciones quirúrgicas y los resultados de las intervenciones, a la luz de las principales publicaciones hasta 2013. Historia
A Etienne-Louis Arthur Fallot le debemos el nombre de una de las malformaciones congénitas del corazón más conocida. Fallot en 18883 hizo una fina descripción de las cuatro (de donde viene el término tetra) características morfológicas básicas, unificándolas en una misma entidad que él llamó la malaide bleue , enfermedad azul, y llevó a cabo la correlación anatomoclínica de tres casos, en el Marseille Medical Journal; sin embargo, la utilización del epónimo Fallot, se atribuye a Maude Abbot en 1924, canadiense, autora también de un excelente atlas sobre cardiopatías congénitas comparadas.4 No obstante, debemos señalar que el autor de la primera descripción conocida de la malformación fue el danés Niels Stensen en 16735 y la primera bella descripción ilustrada correspondió a William Hunter, en Londres, en 1784.6
Definición
Aunque no se han modificado las cuatro características anatómicas fundamentales: defecto septal ventricular, cabalgamiento de la aorta, obstrucción al tracto de salida del ventrículo derecho e hipertrofia ventricular derecha, el foco de la definición, de acuerdo con Anderson,7 se centra en el desplazamiento céfalo anterior del septum infundibular, de la cual deriva la obstrucción al tracto de salida del ventrículo derecho. Van Praagh8 propone que la tetralogía de Fallot es el resultado del subdesarrollo del infundíbulo subpulmonar.
Alcance
Para fines de esta revisión, nos limitaremos al estudio de la forma más frecuente de la tetralogía de Fallot, esto es con análisis segmentario anatómicamente normal sin atresia pulmonar y sin otras anomalías intracardiacas como el defecto septal atrioventricular.
Incidencia, prevalencia y genética
Aproximadamente el 3.5% de los niños que nacen con cardiopatía congénita, tienen TOF, lo que corresponde a un caso por cada 3,600 nacidos vivos o una tasa de 0.28 por cada 1,000 nacidos vivos.9 Sin embargo, el porcentaje sobre el total de cardiopatías congénitas aumenta después del año y alcanza el 10% por la pérdida de enfermos con patologías más graves.
Como ocurre con la mayoría de las cardiopatías congénitas, la etiología precisa de la malformación se desconoce. La mayoría de los casos son esporádicos; sin embargo, se ha reconocido que la microdeleción de la región q11 del cromosoma 22 se presenta hasta en el 25% de los enfermos.10 Estos autores recomiendan que en todo paciente en el que se haga el diagnóstico se realice la búsqueda de la microdeleción mediante FISH.8 El riesgo de recurrencia en el siguiente embarazo se ha estimado en un 3%; adicionalmente, cuando la madre tiene TOF, el riesgo sobre el producto de un embarazo es aproximadamente del 10%; sin embargo, este riesgo es para todas las cardiopatías, lo cual implica que muchas de ellas son menores y no requieren intervención. Lo interesante es que se han reportado algunos casos de gemelos homocigóticos con TOF.11
Diagnóstico
La presentación clínica, depende fundamentalmente del grado de obstrucción pulmonar. La cianosis puede estar atenuada por la presencia de un conducto arteriosos permeable. Cuando la obstrucción es severa y se ha cerrado el conducto, las manifestaciones de hipoxia y acidosis importante se presentan desde las primeras horas o días de vida. Afortunadamente, la mayoría de los niños con TOF se encuentran con mínima cianosis o sin cianosis al nacimiento, pero puede auscultarse un soplo expulsivo pulmonar. Con el paso de días o algunas semanas, la estenosis infundibular se acentúa, la cianosis aparece y eventualmente desencadena crisis de hipoxia.
El diagnóstico definitivo se hace mediante el ecocardiograma transtorácico. Antes de analizarlo mencionaremos los estudios clásicos, a saber: las pistas en la radiografía de tórax son el flujo pulmonar disminuido, la pulmonar excavada, el ápex levantado y particularmente el arco aórtico derecho. Esto se observa en los casos típicos. Es prudente recordar que la tele de tórax puede ser normal en los casos poco severos. En el electrocardiograma se reconoce ritmo sinusal y desviación del eje de QRS a la derecha con hipertrofia ventricular derecha. Clásicamente se observa cambio brusco de V1 a V2, pero no siempre.
El ecocardiograma es el gold standard y debe realizarse de forma urgente, aun si no se cuenta con la placa de tórax o el electrocardiograma. Se recomienda seguir el abordaje subcostal con el análisis segmentario en todos los casos. Una vez establecido el situs solitus y las conexiones concordantes, la visualización de la anatomía intracardiaca permite observar la obstrucción del tracto de salida por la desviación céfalo anterior izquierda del septum infundibular (Figura 1) además de la comunicación interventricular, el cabalgamiento de la aorta, la hipertrofia ventricular derecha y si existen otras lesiones asociadas. Es importante observar el origen de las coronarias en eje corto paraesternal alto para descartar orígenes anómalos. La medición del calibre de las arterias pulmonares es necesaria si se piensa en una paliación con fístula, aunque como veremos más adelante, existe una tendencia a abandonar la fístula por la corrección total. Mediante el abordaje supraesternal se determina la dirección del arco aórtico; es posible, con múltiples cortes, observar colaterales grandes si están presentes.
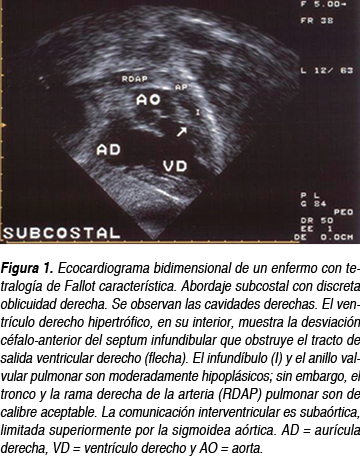
La resonancia magnética y el cateterismo cardiaco (Figura 2) actualmente se usan poco en los casos típicos y el enfermo puede operarse sin ellos (más adelante veremos el estado actual de las indicaciones); cuando se sospechan dañod colaterales, estenosis periférica de ramas, anomalías coronarias o existe alguna duda razonable, se emplea la resonancia magnética o el cateterismo cardiaco, dependiendo de los recursos con los que cuente el centro.
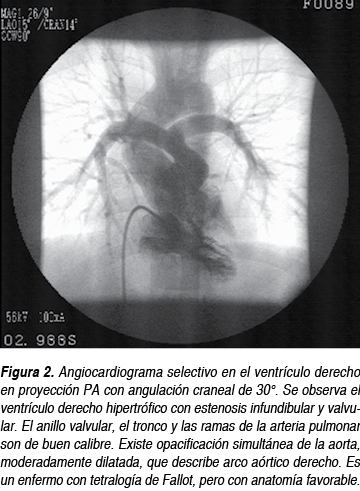
Historia natural y manejo
Sin intervención quirúrgica, la sobrevivencia es pobre y está inversamente relacionada al grado de obstrucción pulmonar. Aproximadamente el 50% de los enfermos que nacen con TOF mueren en los primeros años de vida y difícilmente alguno sobrevive más allá de los 30 años.12 En contraste, en la actualidad puede esperarse que el 90% de los niños operados de corrección total sobrevivan hasta la quinta década de la vida.13 Analicemos por etapas los progresos y las controversias del manejo.
Cirugía paliativa inicial versus cirugía correctiva desde el principio
Desde la primera intervención correctiva con circulación extracorpórea cruzada con un donador (habitualmente uno de los padres) realizada por Lillehei y sus colegas en 1954,14 la tendencia ha evolucionado al realizar la corrección total a edades más tempranas y al abandonar la fístula de Blalock-Taussig original o modificada como paliación. La mortalidad temprana de la corrección total reportada por centros especializados de los países desarrollados ha sido menor al 3%;15 sin embargo, diversos hospitales, en particular en países en desarrollo, mantienen la estrategia de hacer una fístula en el primer año de vida o emplear la cardiología intervencionista con balón o stent como paliación y en un segundo tiempo efectuar la corrección total.16 Es muy interesante, en este sentido, analizar la experiencia del Hospital Sick Children's de Toronto; en su experiencia inicial hacían la corrección en dos tiempos, luego hubo un periodo de transición de 1993 a 1998 en donde fueron abandonando la paliación, de tal modo que el antecedente de fístula antes de corrección total, pasó de 38 al 0% en una serie de 227 casos consecutivos sometidos a corrección total, con una mortalidad de sólo 2.6% y de 0% en los últimos tres años.9 Una revisión multicéntrica de algunos de los mejores centros relativamente recientes reveló que la corrección total puede realizarse incluso antes de los seis meses de edad y sin aumento de la mortalidad. Sólo cuando la operación se realiza en menores de tres meses de vida, se demostró que los días de estancia en la terapia intensiva, el tiempo de intubación endotraqueal y el uso de inotrópicos se encuentran incrementados.17 Es interesante que en el estudio de Van Arsdel y colaboradores concluyen que la mejor sobrevivencia y los mejores resultados fisiológicos se obtienen cuando la corrección total se hace dentro del rango de los 3 a los 11 meses de vida, ni antes ni después.15 Desde luego que hay críticos de la corrección total en edades tempranas; sus argumentos son los siguientes: los efectos adversos de la circulación extracorpórea sobre el cerebro del bebé y la frecuente prolongada recuperación postoperatoria de los enfermos.18 En contraste, las desventajas de plan con dos operaciones son fundamentalmente las siguientes: el enfermo se encuentra expuesto a hipoxia durante mayor tiempo, esto favorece la degeneración de los miocitos y la fibrosis intersticial, lo que a largo plazo favorece las arritmias y la disfunción ventricular y desde luego la exposición del enfermo a dos riesgos operatorios.19
Situación actual en un país en desarrollo
Ahora bien, desde una perspectiva más amplia la posición de un centro en particular sobre la decisión de hacer la corrección en uno o dos tiempos también gravitará, como bien sabemos, dependiendo del entorno y experiencia de ese centro. Con el propósito de conocer los criterios en un país en desarrollo como México, nos dimos a la tarea de entrevistar a los cirujanos o cardiólogos pediatras de cuatro de los principales hospitales que hacen cirugía de congénitos en esta república; ellos son el Dr. Alexis Palacios Macedo, jefe de cirugía cardiovascular del Instituto Nacional de Pediatría; el Dr. Alejandro Bolio, jefe de cirugía cardiovascular Hospital Infantil de México; la Dra. Luisa Beirana, cardióloga pediatra del Hospital de Pediatría del Centro Médico Nacional Siglo XXI, y el Dr. Juan Gómez Vargas, cardiólogo pediatra del Centro Médico de Occidente. En general, la estrategia en estos hospitales es la siguiente: en menores de un año, con crisis de hipoxia o franca desaturación, se hace fístula y corrección total, alrededor del tercer año. Ahora bien, los enfermos estables con poca cianosis son programados para corrección total después del primer año de vida, alrededor del segundo año de edad. Es relevante el hecho de que por lo menos la mitad de los enfermos con TOF llega al hospital tardíamente, después de los tres años de vida. Situación compartida con otros países del mundo en desarrollo.
Seguimiento a mediano y largo plazo
Aunque la mortalidad operatoria de la corrección total inicial de la tetralogía de Fallot ha disminuido de manera constante en estas cuatro décadas hasta lograr series con menos del 3%,15,20 y la sobrevivencia a largo plazo permite que la mayoría viva hasta la quinta década, no se han podido reducir las metas a largo plazo. Primero, el riesgo de muerte tardía, si bien bajó, se mantiene en 0.5 ± 0.07 por año y el riesgo de reoperación a largo plazo se ha mantenido prácticamente sin cambios; el 50% de los enfermos ha sido reoperado después de 30 años.13 Las principales causas de la reoperación son la insuficiencia pulmonar, la estenosis y la comunicación interventricular residuales. Es interesante, en general, que el riesgo de reintervención disminuye con el tiempo: de 2% por año a los 10 años, se redujo a 1.6% por año a los 40 años; sin embargo, el riesgo de muerte se incrementa ligeramente debido a las arritmias y la muerte súbita.13
Sustitución valvular pulmonar
Hace 20 años, todavía se consideraba que la insuficiencia pulmonar posterior a la corrección total era un fenómeno inevitable, universal y bien tolerado a largo plazo. Sin embargo, estudios posteriores demostraron que éste no es el caso. La insuficiencia pulmonar importante se observó relacionada al ensanchamiento progresivo del QRS, a la presencia de arritmias graves, falla ventricular derecha y muerte.21-23 Entre los factores de riesgo asociados a insuficiencia pulmonar a largo plazo, el más importante ha sido el uso de parche transanular al momento de la reparación total.24-26 Sin embargo, no todos están de acuerdo en que el parche transanular sea un factor significativo.13,27 Cuando se comparó, en una misma serie, los Fallot corregidos sin parche transanular contra aquellos que sí lo requirieron, el porcentaje de reemplazo valvular a 20 años fue de 8 y 21% respectivamente.13
Cuándo indicar la sustitución valvular pulmonar
La indicación no es fácil, ya que la mayoría de los enfermos se encuentran asintomáticos aun con insuficiencias significativas. En el año 2000, Therrien y colaboradores28 alertaron a la comunidad científica en este campo. Después de estudiar 25 casos consecutivos de reemplazo valvular pulmonar, concluyeron que las operaciones se habían realizado demasiado tarde; el implante valvular no mejoró los volúmenes ventriculares ni la fracción de expulsión derecha. En un estudio posterior del mismo grupo de investigadores29 en enfermos con menos dilatación ventricular y más jóvenes, encontraron que los límites superiores medidos por la resonancia magnética para obtener regresión del deterioro ventricular derecho eran 170 mL/m2 de volumen diastólico final y 85 mL/m2 del sistólico final. La conclusión es que después de cierto grado de dilatación ventricular derecha, la recuperación de la función es poco probable. Ahora bien, el riesgo operatorio de la sustitución valvular pulmonar es bajo, menos del 1%, y la sobrevivencia estimada a 10 y 20 años es satisfactoria, de 96 ± 2% y 94 ± 3% respectivamente.13 Desafortunadamente, muchos requerirán una reintervención si especialmente, como es lo habitual, se usa prótesis biológica. Para el lector interesado, una revisión exhaustiva sobre las indicaciones y el momento del reemplazo pulmonar en estos enfermos fue realizada por Tal Geva.30
Prótesis pulmonar implantada de forma percutánea
El implante percutáneo de válvulas cardíacas se ha convertido en uno de los campos más fascinantes de la cardiología actual. El implante de la pulmonar es una alternativa real al problema de largo plazo de los enfermos reparados de TOF. A la fecha, en el mundo, más de 700 enfermos han recibido una prótesis pulmonar con ésta técnica, con resultados iniciales alentadores. En la serie más grande publicada con 155 enfermos tratados, no hubo mortalidad periprocedimiento y la mortalidad tardía ha sido muy baja. Los porcentajes libres de reintervención fueron 93, 86, 84, y 70% a 10, 30, 50 y 70 meses respectivamente.31 Una limitación importante es el diámetro del tracto de salida. Cuando éste se encuentra muy dilatado, lo cual es común con la utilización de parche transanular, no se puede implantar la prótesis de forma percutánea.
De cualquier forma, es importante señalar que el implante pulmonar reduce significativamente las arritmias.32
Otras causas de reoperación
En la serie grande de Hickey y colaboradores, la reconstrucción del tracto de salida por estenosis residual o reestenosis ocurrió en un 8% mientras que la ampliación quirúrgica de arterias pulmonares, la reparación de aneurisma de parche del tracto de salida y el cierre de comunicación interventricular (CIV) se hizo en el 9, y 6% respectivamente a 20 años de seguimiento.13 En contraste, la serie del grupo alemán tuvo como primera causa de reintervención el cierre de la CIV en casi la mitad de sus casos reoperados.33 En esta serie, el tiempo promedio transcurrido de la corrección total a la primera reintervención fue de 12.8 años. El reemplazo valvular aórtico por insuficiencia aórtica si es que ocurre, pero es menos frecuente, se realizó en 13 de 973 casos a 20 años.13
Las arritmias a largo plazo
Después de 35 años de la corrección total, el porcentaje de taquicardia ventricular sostenida puede llegar al 11.9% y la muerte súbita al 6%, siendo éste el mecanismo por el que muere un tercio de los casos en etapa tardía19 El substrato más común para ambas es la presencia de dilatación y disfunción ventricular derecha, asociadas o no a insuficiencia pulmonar importante, pero vinculadas a ensanchamiento del QRS ≥ 180 ms del electrocardiograma. Como se dijo antes, el implante pulmonar reduce el riesgo de arritmias al reducir el tamaño del ventrículo derecho, siempre y cuando la intervención haya sido oportuna. Frecuentemente, el manejo de las arritmias graves consiste en un tratamiento combinado, a saber: antiarrítmicos y ablación durante el estudio electrofisiológico o mapeo y crioablación quirúrgica. Con estas combinaciones, la mayoría de los enfermos mejoran sin embargo, puede ser necesario el implante quirúrgico de un desfibrilador. Los enfermos postoperados de TOF ocupan el primer lugar en el uso de desfibrilador dentro del grupo de las cardiopatías congénitas.34,35 Seguimiento
Si bien la expectativa de vida de los enfermos operados de TOF es muy buena, alrededor de la mitad requerirá por lo menos una reintervención a lo largo de su vida, de modo que no son enfermos curados y todos deben ser evaluados periódicamente de por vida. Para los casos sin lesiones residuales significativas ni arritmias, lo recomendable es una visita anual con valoración clínica, eléctrica, radiológica y ecocardiográfica. En los enfermos con obstrucción residual poco importante en el tracto de salida del ventrículo derecho, debe medirse con ECO Doppler el gradiente por lo menos cada seis meses, un gradiente igual o mayor a 50 mmHg es una indicación de intervención; cuando existe insuficiencia pulmonar ésta debe evaluarse con ecocardiografía y resonancia magnética para medir función ventricular y volúmenes ventriculares; los umbrales para intervenir son 170 mL/m2 de volumen diastólico final y 85 mL/m2 de volumen sistólico en el ventrículo derecho. La tomografía computada puede ser la alternativa en los casos con marcapaso o desfibrilador implantado. La prueba de esfuerzo y el monitoreo de Holter son las herramientas iniciales en presencia de extrasístoles frecuentes o arritmias. Es competencia del especialista en ellas el estudio electrofisiólogico y la posible ablación. La comunicación interventricular residual puede ser tolerada si no produce dilatación ventricular ni hipertensión arterial, de lo contrario puede considerarse el cierre percutáneo o quirúrgico. En cada cita es importante evaluar la función de la válvula aórtica. Toda reintervención debe ser discutida y planeada en conjunto con el cirujano. Los enfermos con lesiones residuales aún poco significativas, tienen riesgo de endocarditis bacteriana. En ellos está indicada la profilaxis con antibióticos.
Embarazo
Todas las pacientes enfermas operadas con TOF, antes de embarazarse, deberían ser sometidas a una valoración integral que incluya al especialista en cardiopatías congénitas en el adulto, un cardiólogo especialista en arritmias y el gineco-obstetra. El riesgo del embarazo depende del estado hemodinámico de las enfermas. Aquellas asintomáticas y sin lesiones residuales, podrán tener un embarazo y un parto por vía vaginal con riesgo equivalente a la población normal; sin embargo, las pacientes enfermas con insuficiencia pulmonar significativa, dilatación del ventrículo derecho y arritmias, tienen tanto ellas como sus productos, debido a la sobrecarga de volumen que impone el embarazo.36,37 En estos casos es conveniente considerar los beneficios del implante valvular pulmonar. Si la enferma es vista cuando ya está embarazada, situación no poco frecuente, debe evaluarse de manera conjunta la operación cesárea aun antes del término, dependiendo de las manifestaciones clínicas. En ellas, la posibilidad del implante percutáneo es una opción atractiva.
El futuro próximo
Ya está en curso un proyecto de investigación en el que participan cuatro de los principales centros del mundo (el Children's de Boston, Cardiac Center de Toronto , Royal Brompton and Harefield NHS Foundation de Londres y el Academic Medical Center de Amsterdam ), enfocados a identificar los factores de riesgo para taquicardia ventricular sostenida y muerte en los enfermos postoperados de corrección de Fallot en la infancia, utilizando la clínica, el electrocardiograma y la resonancia magnética;38 a la fecha de su publicación preliminar, han reunido 873 pacientes. Este estudio aportará conocimiento nuevo sobre factores de riesgo que puedan ser evitados con oportunidad.
Sin duda la prótesis implantable de manera percutánea, se perfeccionará. A partir del uso de células autólogas progenitoras, se han podido desarrollar válvulas cardiacas humanas e implantarse en dos enfermos.39 Después de poco más de tres años, los resultados son satisfactorios. La expectativa es prometedora; se trata de lograr una válvula que crezca con el enfermo y que no se deteriore a largo plazo. Mientras tanto, la ecocardiografía fetal afina con más detalle el diagnóstico prenatal de la TOF40 y las técnicas quirúrgicas y el manejo postoperatorio se superan, logrando que cada vez en más hospitales se obtengan sobrevivencias cercanas a 100%.
Agradecimientos
A los doctores Alejandro Bolio, Alexis Palacios Macedo, Juan Gómez y Luisa Beirana por sus valiosas opiniones, al PhD. Héctor Alva por su apoyo con el idioma inglés y a la Lic. Sabimel Rendón por su apoyo bibliográfico.
REFERENCIAS
1. Blalock A, Taussig HB. Surgical treatment of malformations of the heart; in which there is pulmonary stenosis or atresia. JAMA. 1945; 128: 189-202. [ Links ]
2. Taussig HB. Neuhauser lecture-tetralogía de Fallot: early history a late results. Am J Roentgenol. 2009; 133: 423-431. [ Links ]
3. Fallot A. Contribution a l'anatomie pathologique de la maladie bleue (cyanose cardiaque). Marseille Medicale. 1888: 71-93, 138-158, 207-223, 270-286 y 341-354. [ Links ]
4. Abbott ME. Atlas of congenital cardiac disease. New York: American Heart Association; 1936. [ Links ]
5. Stensen N. Embryo monstro affinis Parisiis dissectus. Acta medica et philosophica Hafniencia 1673; 1: 202-203. In: Maar V, Stenonis Nicolai. Opera philosophica. Copenhagen. 1910; 2: 49-53. [ Links ]
6. Hunter W. Three cases of malformation of the heart. In: Cadell T. Medical observations and inquiries, vol. 6. Society of Physicians in London; 1784. [ Links ]
7. Anderson RH, Jacobs ML. The anatomy of tetralogy of Fallot with pulmonary stenosis. Cardiol Young. 2008; 18: 12-21. [ Links ]
8. Van Praag R. The First Stella Van Praagh memorial lecture: the history and anatomy of tetralogy of Fallot. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2009; 12: 19-38. [ Links ]
9. Apitz C, Weeb GD, Redington AN. Tetralogía de Fallot. Lancet. 2009; 374: 1462-1471. [ Links ]
10. Weber SA, Hatchell EI, Barber JCK et al. Importance of microdeletions of chromosomal región 22q11 as a cause of selected malformations of the ventricular outflow tracts and aortic arch: a three-year prospective study. Pediatrics. 1996; 129: 26-32. [ Links ]
11. Alva C, Gómez FD, Jiménez-Arteaga S et al. Concordance of congenital heart defects in two pairs of monozygotic twins: pulmonary stenosis and tetralogy of Fallot. Arch Cardiol Mex. 2010: 80: 29-32. [ Links ]
12. Bertranou EG, Blackstone EH, Hazelrig JB et al. Life expectancy without surgery in tetralogy of Fallot. Am J Cardiol. 1978; 42: 458-466. [ Links ]
13. Hickey E, Veldtman G, Bradley TJ et al. Late risk of outcomes for adults with repaired tetralogy of Fallot from an inception cohort spanning four decades. Eur J Cardiothorac Surg. 2009; 35: 156-166. [ Links ]
14. Lillehei CW, Cohen M, Warden HE et al. Direct vision intracardiac surgical correction of the tetralogy of Fallot, pentalogy of Fallot and pulmonary atresia defects; report of first ten cases. Ann Surg. 1955; 142: 418-442. [ Links ]
15. Van Arsdell GS, Maharaj GS, Tom J et al. What is the optimal age for repair or tetralogy of Fallot? Circulation. 2000; 102(19Suppl 3): III123-129. [ Links ]
16. Dohlen G, Chaturvedi RR, Benson LN et al. Stenting of the right ventricular outflow tract in the symptomatic infant in tetralogy of Fallot. Heart. 2009; 95: 142-147. [ Links ]
17. Vohra HA, Adamson L, Haw MP. Is early primary repair for correction of tetralogy of Fallot comparable to surgery after 6 months of age? Interact Cardiovasc Thorac Surg. 2008; 7: 698-701. [ Links ]
18. Zeltser I, Jarvik GP, Bernbaum J. Genetic factors are important determinants of neurodevelopmental outcome after repair of tetralogy of Fallot. J Thorac Cardiovasc Surg. 2008; 135: 91-97. [ Links ]
19. Venugopal P. Histopathology of the right ventricle outflow tract and its relationship to clinical outcomes and arrhythmias in patients with tetralogy of Fallot. J Thorac Cardiovasc Surg. 2006; 132: 270-277. [ Links ]
20. Alexious C, Mahmoud H, Al-Kahaddour A et al. Outcome after repair of tetralogy of Fallot in the first year of life. Ann Thorac Surg. 2001; 71: 494-500. [ Links ]
21. Gatzoulis MA, Balaji S, Webber SA et al. Risk factors for arrhythmia and sudden death late after repair of tetralogy of Fallot: a multicenter study. Lancet. 2000; 356: 975-981. [ Links ]
22. Roos-Hesselink J, Perlroth MGJ, Spitaels S. Atrial arrhythmias in adults after repair of tetralogy of Fallot. Correlations with clinical, exercise, and echocardiographic findings. Circulation. 1995; 91: 2214-2219. [ Links ]
23. Harrison DA, Siu SC, Hussain F et al. Sustained atrial arrhythmias in adults late after repair of tetralogy of Fallot. Am J Cardiol. 2001; 87: 584-588. [ Links ]
24. Hokanson JS, Moller JH. Adults with tetralogy of Fallot: long-term follow up. Cardiol Rev. 1999; 7: 149-155. [ Links ]
25. Zhao HX, Miller DC, Reitz BA et al. Surgical repair of tetralogy of Fallot. Long term follow up with particular emphasis on late death and reoperation. J Thorac Cardiovasc Surg. 1985; 89: 204-220. [ Links ]
26. Lim C, Lee JY, Kim VH et al. Early replacement of pulmonary valve after repair of tetralogy: is it really beneficial? Eur J Cardiothorac Surg. 2004; 25: 728-734. [ Links ]
27. d'Udekem Y, Ovaert C, Grandjean F et al.Tetralogy of Fallot: transannular and right ventricular patching equally affect late functional status. Circulation. 2000; 102: III116-122. [ Links ]
28. Therrien J, Siu SC, McLaughhlin PR et al. Pulmonary valve replacement in adults late after repair of tetralogy of Fallot: are we operating too late? J Am Coll Cardiol. 2000; 36: 1670-1675. [ Links ]
29. Therrien J, Provost Y, Merchant N et al. Optimal timing for pulmonary valve replacement in adults after tetralogy of Fallot repair. Am J Cardiol. 2005; 95: 779-782. [ Links ]
30. Geva T. Indications a timing of pulmonary valve replacement after tetralogy of Fallot repair. Semin Thorac Cardiovasc Surg Pediatr Card Surg Ann. 2006; 9: 11-22. [ Links ]
31. Lurz P, Coats L, Khambadkone S et al. Percutaneous pulmonary valve implantation: impact of evolving technology and learning curve on clinical outcome. Circulation. 2008; 117: 1964-1972. [ Links ]
32. Therrien J, Siu SC, Harris L et al. Impact of pulmonary valve replacement on arrhythmia propensity late after repair of tetralogy of Fallot. Circulation. 2001; 103: 2489-2494. [ Links ]
33. Tirilomis T, Friedrich M, Zenker D et al. Indications for reoperation late after correction of tetralogy of Fallot. Cardiol Young. 2010; 20: 396-401. [ Links ]
34. Khairy P, HarrisL, Landzberg M et al. Implantable cardioverter-defibrillators in tetralogy of Fallot. Circulation 2008;117:363-370. [ Links ]
35. Yap SC, Roos-Hesselink JW, Hoendermis ES et al. Outcome of implantable cardioverter defibrillators in adults with congenital heart disease: a multicenter study. Eur Heart J. 2007; 28: 1854-1861. [ Links ]
36. Torres-Gómez L, Iñigo-Riesgo CA, Espinosa-Ortegón MA et al. Embarazo y tetralogía de Fallot con y sin corrección quirúrgica. Ginecol Obstet Mex. 2010; 78: 309-315. [ Links ]
37. Veldtman GR, CoOnnolly HM, Grogan M et al. Outcomes of pregnancy in women with tetralogy of Fallot. J Am Coll Cardiol. 2004; 44: 174-180. [ Links ]
38. Valente AM, Gauvreau K, Assenza GE et al. Rationale and design of an international multicenter registry of patients with repaired tetralogy of Fallot to define risk factors for late adverse outcomes: The INDICATOR Cohort. Pediatr Cardiol. 2013; 34: 95-104. [ Links ]
39. Ceborati S, Lichtenberg A, Tudorache I et al. Clinical application of tissue engineered human heart valves using autologous progenitor cells. Circulation. 2006; 114(Suppl 1): 132-137. [ Links ]
40. Bailliard F, Anderson RH. Tetralogy of Fallot. Orphanet J Rare Dis. 2009; 4: 2. [ Links ]

