











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Models are essential in education; chemical education requires several model systems to discuss complicated topics. Models are never completely true, but they can be useful if they can help students to understand a topic. When a scientific model is not based on facts its utility is voided; for any reason or limitation, its removal from use must be warranted. We propose in this analysis, with examples to show major problems in their continued use, that the models of hybrid atomic orbitals (HAO) and valence-shell electron-pair repulsion (VSEPR) are outmoded. The purpose of this article is to give clear evidence of the misconceptions that arise because of the (mis)use of HAO and VSEPR in introductory chemistry courses; in a second article we provide solutions of this problem.
Of previous critiques of the hybrid-atomic-orbital (HAO) model in organic chemistry (Lamoureux & Ogilvie, 2019a; Lamoureux & Ogilvie, 2019b), the first delineated six logical fallacies about hybridization; the second revealed seven practical problems with the use of HAO in organic chemistry. Because of limited space and the complicated subject matter, a thorough discussion of the pedagogical issues involved in teaching HAO to introductory students was impracticable. Here we review the problems with when and where to teach HAO in introductory chemistry and the inconsistent use of HAO for systems of every type. Without a complete analysis of the use of HAO in chemistry, students will continue to be confused, misled and frustrated. In the twenty-first century, with HAO considered dogma in most textbooks, the continuation of this flawed model is unacceptable.
VSEPR, originated by Sidgwick and Powell (Sidgwick & Powell, 1940) and extended by Gillespie (Gillespie & Nyholm, 1957), provides a model system to predict the three-dimensional structure from a Lewis structure. Despite severe criticism of VSEPR in the 1970s (Drago, 1973) this model continued to dominate teaching chemistry without dissent for almost 50 years. Some problems with VSEPR are discussed below, especially its incompatibility with other models. We provide several concrete examples of these difficulties, which cause confusion to many students.
When and Where to Use Hybrid Atomic Orbitals
In assessing a student’s knowledge of hybrids and hybridization, most instructors apply a diagnostic test. Representative questions ask when and where to use hybridization. Hybridization of three types (sp 3 /sp 2 /sp, tau and non-integral) is in use in introductory chemistry; the possibility of no hybridization is sometimes invoked (Lamoureux & Ogilvie, 2019b). If instructors and textbooks cannot agree on how, when and where hybridization applies, the students receive conflicting signals and become chronically confused.
Hypervalent Main-group Elements
What is the evidence of hybridization for hypervalent molecules (e.g. SF6, PCl5, XeF6, IF7)? Using d 2 sp 3 hybridization to ‘explain’ the shape or bonding in SF6 was criticized unequivocally Coulson (by Coulson, 1969), Magnusson (Magnusson, 1990) and Cooper et al. (Cooper et al., 1994) many years ago. Gilheany rigorously evaluated the evidence for phosphorus compounds and concluded that hybridization involving d orbitals did not exist (Gilheany, 1994). These critiques were widely accepted but hybridization continued to be taught in classrooms because it was a simple model. “The model of spd hybridization to ‘explain’ the bonding in hypervalent compounds of heavier main-group elements is an example. Several quantum-chemical studies have clearly shown that the d orbitals of the heavier main-group elements are not really engaged as valence orbitals in chemical bonding but rather serve as polarization functions.” This condition is acknowledged in the latest edition of a popular textbook of inorganic chemistry, but it then continues “nevertheless, the concept of hybrid orbitals retains advantages of simplicity and, in many instances, affords a very easy way to correlate and ‘explain’ molecular structures. The danger in applying correlations as pseudo-explanations lies in the temptation to take good correlations as a proof for the existence of the underlying assumption” (Frenking & Fröhlich, 2000).
In a recent summary of the use of quantum-chemical calculations to determine the possibility of spd hybridization in higher main-group elements, Galbraith concluded: “Although the idea of spd hybridization is simple enough for general-chemistry students to grasp and it works well to describe the bonding in hypervalent molecules, it is fundamentally flawed, and therefore, in the author’s opinion, should be removed from the general chemistry curriculum…In many cases a strict sp, sp 2 , or sp 3 hybridization scheme is inadequate even among molecules containing second-period atoms…Hybridization was a good theory based on the experimental evidence of the day. The role of d orbitals in other hypervalent molecules is expected to be similar, thus eliminating the need for spd hybridization in any form.” (Galbraith, 2007).
Recent textbooks are finally removing the insistence of applying hybridization to all molecules. “Hybridization is not a real phenomenon, but an after-the-fact rationalization of an experimentally determined result. … theoretical considerations cast serious doubt on d-electron participation. The same doubt, of course, extends to the use of d orbitals in hybridization schemes. Despite the difficulty posed by hybridization schemes involving d orbitals, the sp, sp 2 and sp 3 hybridization schemes are well established and commonly encountered, particularly among the second-period elements.” (Petrucci et al., 2017). Many textbooks, and many instructors, continue to use hybridization in hypervalent cases even in the face of all this evidence to the contrary.
Other Main-group Elements
Main-group elements in the third period without hypervalency (e.g. SiH4, PH3, H2S) have been examined for evidence of hybridization. Kutzelnigg presented convincing arguments against hybridization: “Many concepts that have been justified for first-row elements (Li to Ne) cannot - contrary to widespread belief - be generalized to the higher main-group elements. This applies particularly to the concept of hybridization, which should be viewed with considerable caution.” (Kutzelnigg, 1984). Frenking added that “the model of hybridization already becomes less straightforward for heavier atoms than the first-row elements C-Ne”(Frenking & Fröhlich, 2000). As an example, SiH4 was shown not to have sp 3 hybridization, because of inefficient overlap between 3s and 3p orbitals on Si (Frenking & Fröhlich, 2000). A recent textbook described the bonding in H2S and PH3 without hybridization (Petrucci et al., 2017).
To answer the question posed at the beginning of this section, it seems that hybridization has been used wherever and whenever possible in chemistry, but overwhelming evidence shows that most textbooks teach HAO using only select molecules. The only agreement seems to be that HAO apply mainly to second-period elements, and to organic chemistry in general.
Can One Apply Hybridization to Everything?
Molecules
A most perplexing misconception, in a pedagogical sense, is the application of HAO to all aspects of molecular structure. It is completely unacceptable to apply hybridization to an entire molecule, but in one survey several students responded positively to a query that “methane is a sp 3 hybridization state” (Salah & Dumon, 2011). Once hybridization is applied to molecules, it becomes a slippery slope to confuse what the term means; this trend is unfortunately increasing. For example, for a recent research article “An sp-Hybridized Molecular Carbon Allotrope” (Kaiser et al., 2019), the title gains brevity only with the sacrifice of comprehension. Confusion arises because that designation sp for hybridized orbitals is simply an artefact of orbitals from the solution of the Schroedinger equation in spherical polar coordinates; the same shape and extent of orbitals arises from an analogous solution in paraboloidal coordinates (Ogilvie, 2016), completely undermining the postulate of HAO.
‘Atoms in Molecules’
One common query in evaluations is to identify the hybridization of the selected atoms in a particular molecule. This query is nonsensical within the framework of hybridization for two reasons. First, ‘atoms in molecules’ cannot ‘have hybridization’. In fact, there is no atom in a molecule in quantum mechanics; any attempt to define an atom within a molecule involves an arbitrary criterion. One can speak of atomic centers (associated with atomic nuclei) in a molecule, which, except for hydrogen, might resemble the distribution of electronic charge density of isolated or free atoms, but the only objectively recognizable constituents of a molecule are atomic nuclei and their associated environment of electronic charge density. “When atoms form ions or interact to form molecules, the number and/or the specific structural arrangement of these electrons is destroyed. Indeed, this electronic rearrangement is the very essence of chemical change, and it follows as a necessary corollary of this fact that neutral atoms cannot serve as structural units within polyatomic molecules and ions. As Mulliken and other proponents of molecular-orbital theory have repeatedly emphasized over the last 60 years, molecules are made of nuclei and electrons and not of neutral atoms” (Jensen, 1998).
Second, in a process of preparing for calculations to describe the formation of molecules, atomic orbitals of each atom become collected in combinations to form molecular orbitals; there should be no trace of the previous states in the final state. A most common confusion among students is the inability to distinguish among atomic, hybrid and molecular orbitals (Taber, 2002). This confusion extends also to most textbooks. HAO do not exist in molecules - they have no physical existence anywhere - they are merely artefacts, in the form of algebraic formulae, within calculations according to one particular method of quantum mechanics, even though almost all textbooks indicate HAO in molecules with arrows pointing to atomic centers. Only one textbook that we could find clearly disparages this usage of hybridization: “The common practice of referring to a molecule or an atom as ‘rehybridising’ is not good usage - the rehybridisation in question is in our picture, not in the molecule. It is likewise poor (but unfortunately common) practice to refer to atoms as being sp 3 , sp 2 or sp hybridised. Again, the atoms themselves are not hybridised; it is we who have chosen to picture them that way. It is better in such circumstances to refer to the atoms as being tetrahedral, trigonal or digonal” (Fleming, 2011). It is better still to state that the atomic centers in molecule have a tetrahedral, or trigonal, or digonal, disposition of adjacent atomic centers and to eliminate hybridization completely.
Bonds
An equally poor practice is to assign a hybridization to bonds in molecules. “It is also strictly incorrect to define a bond as being formed by the overlap of atomic or hybrid orbitals. This is simply a convenient description of a hypothetical process.” (Gillespie et al., 2001). Covalent bonds are defined by the participation of electrons shared among nuclei, although the extent of such sharing might be slight as we have noted above. There is no HAO present in a bond, by definition. A recent quantum-chemical study using the spin-coupled general valence-bond (SCGVB) method tried to answer this question: How are atoms changed on formation of a molecule? (Dunning et al., 2016). Only a trace of s-p orbital hybridization could be identified in the retro-analysis of the bonding of molecules ethyne, ethene or methane (Xu & Dunning, 2020). These authors concluded that “These findings imply that, in modern valence-bond theories, other factors are responsible for the structures and properties of molecules that are traditionally attributed to orbital hybridization” (Xu & Dunning, 2020). They summarized the results in a 2021 paper: “However, the VBSCF and SCGVB orbitals are not the sp 3 , sp 2 and sp hybrid orbitals of traditional VB theory…But, what about the use of sp 3 orbitals to define the tetravalence of carbon and the tetrahedral geometry of methane? The energy lowering associated with 2s−2p hybridization combined with Pauli repulsion between the resulting bond pairs leads, instead, to the observed tetrahedral structure of methane with four equivalent bonds” (Dunning et al., 2021). One must thus not rationalize structures based only upon a traditional bond hybridization. Please, if you value critical thinking, remove all questions asking about the ‘hybridization of bonds’ because the situation is not simple and all structures depend on many energetic factors.
Lone pairs
What battles have been raged over the trivial matter of lone-pair hybridization! Based on the argument presented above, there should be no lingering trace of hybridization for lone pairs in molecules, but this fact has not prevented the attempt. We first clarify the limitations on the hybridization of lone pairs - recall that the justification of hybridization depends on the bond angle among nuclei. Diatomic species (such as CO, CN− etc.) are trivial cases in which there can never be evidence to discuss hybridization because bond angles are defined for exactly three adjacent nuclei. Furthermore, for any terminal atomic center with nominal lone pairs (e.g. -F, -Cl, -Br, -I, -O−, =O, =S, ≡N) it is an exercise in futility to ‘assign’ a hybridization to the lone-pair electrons; how would one confirm or deny the existence of any hybrid without evidence of bond angle?
This first limitation might come as a shock to organic chemists who are accustomed, for example, to draw ‘rabbit ears’ separated by 120° on a carbonyl oxygen: there is neither experimental nor calculational evidence for this geometry. Experimentally, one can measure only the molecular electron density; one cannot assign unique geometrical spaces to four electrons on oxygen into ‘lone pairs’ (Truhlar et al., 2019). Studies of hydrogen bonds to carbonyl compounds show almost random directionality of X-H•••O=C; there is no evidence of highly selective geometry at 120° (Anslyn & Dougherty, 2006). Quantum-chemical calculations can show canonical (delocalized) electrons or the electrons can be localized with several schemes, each providing a different ‘orbital picture’, that are mathematically equivalent (Truhlar et al., 2019). This fact is consistent with a discussion of hybridization schemes of the various types: if no hybridization is applied for oxygen in a carbonyl group, the lone-pair electrons might be considered as derived from pure s and p atomic orbitals; if the tau hybridization model is used, a sp 3 geometry (interorbital angle ~109.5°) of the lone pairs is expected; if the common sp 2 model is used, an interorbital angle 120° is assumed (Figure 1). The assignment becomes even more complicated when a resonance structure (with three purported lone pairs!) is comparatively analyzed. It is the pinnacle of illogic to accept that electron density in a molecule is fluxional, as justified using resonance structures, and then to claim that a lone pair is fixed in space, of a certain size and shape that can be distinguished from all other electrons.
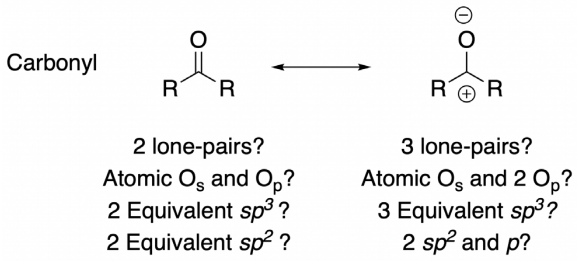
Figure 1: Lewis’s structure of a general carbonyl compound with its corresponding resonance structure and possible hybridization schemes for the purported lone pairs on oxygen in each case.
We proceed to examine the most common case in which hybridization of lone pairs is attempted: dicoordinate oxygen (e.g., water). One should note that Pauling, in his epochal 1931 paper, (Pauling, 1931) favored a no-hybridized model for oxygen. One perspective over 30 years ago, titled ‘No Rabbit Ears on Water’, discussed how to teach hybridization of lone pairs (Laing, 1987). This flawed paper advised the rejection of sp 3 hybridization to describe the bonding in water and suggested use of the no-hybridized model (unfortunately including the statement that this model has experimental evidence - there is none!). This paper was countered by another article recommending teaching the equally flawed ‘squirrel-ear’ model for water (Martin, 1988).
In 2014, several authors combined forces to critique both the VSEPR model and the related sp 3 hybridized lone pairs in water. They applied a quantum-chemical method described as natural bond order (NBO) to present the n(s)-n(p) model of inequivalent lone pairs on oxygen, for not only water but also methanol, formic acid, and furan. They referred to the ‘rabbit-ear’ equivalent lone pairs as anachronisms and not even “proper lone pairs” (Clauss et al., 2014).
This paper by Clauss et al. was refuted the next year in a counter-argument that concluded “It follows from what precedes that one must give up the belief that there exists a unique set of supposedly ‘real’, or ‘best’, orbitals for a given molecule, e.g. the lone pairs of H2O…Thus, each set of lone-pair orbitals has its own preferred domain of application, but they all lead to the same molecular properties and to the same unique electron-density distribution in 3D space” (Hiberty et al., 2015). These authors did not reflect on the teaching of lone pairs, but rather invalidated the assertion that there was only one legitimate lone-pair model. This assertion has strong recent scientific support (Truhlar et al., 2019), but the pedagogical implications have not caught up in recent textbooks.
Subsequently, the comment of Hiberty et al. above was criticized; once again the opinion was stated that the n(s)-n(p) model is considered a better investment of pedagogical effort than that currently devoted to superficial rabbit ears and VSEPR-type rationalizations (Clauss et al., 2015). A plan to teach lone pairs exclusively as the n(s)-n(p) model was then presented in a following article (Esselman & Block, 2018). Unfortunately, the labeling of tetrahedral oxygen electrons in water as “the wrong lone pairs” and the n(s)-n(p) model as the “observable structure” eliminated any scientific respectability of this article, as also the inclusion of this questionable statement: “The incorrect assumption that a divalent oxygen atom with two lone pairs has a tetrahedral electron geometry is one of the more problematic assumptions of VSEPR with respect to its impact on the understanding of organic molecules” (Esselman & Block, 2018).
Beyond the simple cases such as water, the sleight of hand in reassigning hybrids has serious consequences in consistent pedagogy. In the case of cyclic ethers tetrahydrofuran and furan (Figure 2), the Lewis structures are similar; the C-O-C bond angles in these two molecules are almost identical, and VSEPR rules are the same: the oxygen should be AX2E2 (tetrahedral). It is not unexpected, therefore, for students to assign the lone pairs due to the use of sp 3 HAO, with an interorbital angle ~109.5° for both molecules. The situation in the case of cyclic amines pyrrolidine and pyrrole (Figure 2) is similar: analogous Lewis structures and C-N-C angles approximately equal. The N should be AX3E (trigonal pyramidal); the lone pair is expected to show the same sp 3 hybridization for both amines. In both cases, diligent students who apply the memorized rules and who follow logical assumptions are rewarded with a zero in an examination; they are told that these pairs of molecules are not alike and do not share the same hybridization model. Furan and pyrrole are aromatic, which changes the rules. “Often we will see an atom adopt a nonstandard hybridization in order to maximize resonance. This most commonly arises when an atom has a lone pair of electrons that is in conjugation with (directly bonded to) a π system…Such resonance effects on hybridization are common and should be routinely looked for when assigning hybridization to various atoms [sic]” (Anslyn & Dougherty, 2006)..
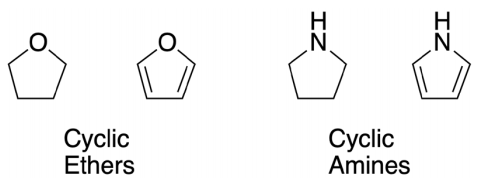
We summarize where one should not use HAO: for entire molecules, for ‘atoms in molecules’, for bonds or for lone pairs. One can always extend the definition to try to encompass these items (e.g. which hybridization on atomic carbon can be used to overlap with atomic hydrogen s orbitals to form ethyne?) but a question of this type in practice is rarely used and lacks pedagogical value. Note that, with our recommendation about the removal of lone pairs on halogens, oxygen and nitrogen, carbon is the only element left that might be considered to need hybridization.
What is Problematic with VSEPR?
Most students learn VSEPR in the first semester to ‘predict’ molecular structure but it becomes a memorization task. “The student is taught (i) to draw a Lewis (electron-dot) structure, (ii) to count the number of ‘electron clouds’ in the Lewis structure, and (iii) to relate that number to the standard VSEPR table of structures and geometries, which can be found in any first-year textbook” (Lindmark, 2010).
The rules of VSEPR are easy to memorize (Gillespie, 2004), but this model is flawed and undermined by the many exceptions to the rules (Myers, 1992). Several tenets of VSEPR, for example that non-bonding electrons occupy a ‘larger’ space than bonding electrons, have been challenged (Clauss et al., 2015).
Beyond the problems of inconsistent rules, VSEPR cannot fulfill its role of a predictive model because it tries to connect a simple (Lewis) line drawing to a much more complicated quantum object. Molecules are three-dimensional and dynamic; nuclei move and electrons flow in a sea of indistinguishability (Laszlo, 2013). Both VSEPR and hybridization are assigned to static 2D structures; this limitation of both models is the cause of many non-standard cases, and a source of consternation for many students.
Is Hybridization Compatible with VSEPR and Resonance?
Hybridization and VSEPR
From its initial development, VSEPR was never considered to be compatible with HAO. It is interesting that in the same article titled the “VSEPR model”, Gillespie then redefined it to be a “VSEPR theory” that is based on quantum mechanics but is somehow separate from it. Gillespie confusingly claimed “[VSEPR] does attempt to provide a simple model for the prediction of molecular geometry which is soundly based on quantum mechanics…It should be noted that the VSEPR theory makes no use of atomic orbitals or of hybrid orbitals formed from these atomic orbitals, and it thus avoids the possibility of misleading a student into thinking that hybrid orbitals give an explanation for molecular geometry when, in fact, they provide nothing more than an approximate description of the bonding when one already knows the geometry” (Gillespie, 1974).
VSEPR is typically taught before HAO and the two models are subsequently linked, but hybridization is later used in a cyclic argument: “The description of the bonds around each carbon atom [in ethene] in terms of sp 2 hybrid orbitals forming σ bonds plus a p orbital forming a π bond is based on the known geometry of the ethene molecule so this description of the bonding cannot be used to predict the molecular geometry.” (Gillespie, 1992).
Resonance and VSEPR
In a preceding section we wrote that even simple molecules such as water have been accorded various mutually exclusive teaching models. How can students assimilate knowledge with many models, especially on exposure to conflicting models from separate textbooks, instructors or courses? We propose that a major logical impediment is the incompatibility of the model of hybridization with the models of VSEPR and resonance. The three systems are incongruous and can cause cognitive dissonance among students.
The following case study of isonitriles is an interesting example wherein a conflict among resonance, VSEPR and hybridization is highlighted. The structure (carbene-like) on the left in Figure 3 has a nitrogen atom with VSEPR assignment AX2E (trigonal planar) and might be described as using sp 2 HAO. The zwitter-ionic resonance structure on the right (which, recall, is another representation of the same molecule) requires VSEPR assignment AX2 (digonal) for nitrogen that is associated with sp hybridization. A difference 60° exists in the predicted R-N-C bond angles between structures. Each separate Lewis structure would have a distinct VSEPR assignment, hybridization and predicted geometry! Of the two assignments which is the correct one? Or does the truth lie somewhere between? Isonitriles are known to be linear (Ramozzi et al., 2012); only by previous knowledge of the three-dimensional structure can one then justify post hoc the preferred selection.
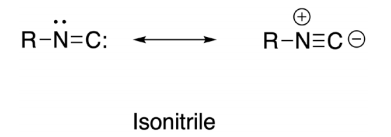
A Tale of Two Molecules
As another instance of the incompatibility of hybridization, VSEPR and resonance in teaching organic chemistry, we show two molecules of which the structure is well known, but one cannot logically use the same models to provide information about them. In Figure 4 are shown the Lewis structures of an amide and an aniline, with possible corresponding resonance structures for comparison. An astute student might ask ‘Is there evidence of the delocalization of the electrons illustrated by the resonance structures?’ Based on recent research, chemists should be skeptical. “Another example is the hybridization of N in an amide, which should be classified as sp 2 . The validity of resonance in an amide has recently come into question.” (Anslyn & Dougherty, 2006). Data from Hammett-Swain parameters (σp = -0.66, R = -0.74) (Hansch et al., 1991) indicate that -NH2 as substituent donates strongly into the π system of the aromatic ring in aniline, but other experimental facts are inconclusive: “Evidence concerning geometries of anilines derived from aqueous basicities can be misleading. Also, the other type of evidence from dipole moments, derived from vector addition of bond dipoles, requires location of electron densities” (Sudlow & Woolf, 1998).
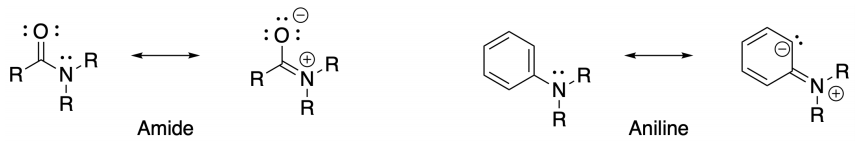
If one accepts the resonance picture, at first glance these structures would appear to have the same influences. Consider nitrogen in both structures: N has three bonds and one lone pair (AX3E in VSEPR vernacular) in the left resonance structure and three electron-pair domains and no lone pair (AX3) in the right resonance structure. There is obviously a logical problem in assigning one geometry according to VSEPR rules. Perhaps the assigned geometry should be intermediate between trigonal pyramidal (AX3E) and trigonal planar (AX3) in both cases? Should one use the same model for both molecules if the Lewis structures appear similar?
The actual geometrical structures are well known. For example, for acetamide, the environment about N is almost planar with N-C=O bond angle 122.0±0.1°; most amides are considered to be planar (Szostak & Aube, 2013). For aniline, N is pyramidal with H-N-H bond angle 113.9±0.3° - in fact it is well known that the calculated and X-ray structures of aniline and substituted anilines are pyramidal (Sudlow & Woolf, 1998). On application of the hybridization model to the known structures, one would assign the N hybrid orbitals as corresponding to sp2 for amides and sp3 for anilines. The similarity in Lewis structures (including resonance) fails to correspond to similarity in the models.
Not all amide nitrogens have a planar environment (Li et al., 2020). Not all aniline nitrogens have a pyramidal environment (Sudlow & Woolf, 1998). The experimental geometry of similar molecules might vary according to the method of determination, phase (solid, liquid or gas), presence of hydrogen bonds or other interactions, or varying substituents (Sudlow & Woolf, 1998). Both student A who memorizes the rules for VSEPR and hybridization to predict sp3 hybridization in amides and student B who memorizes that there is sp 2 hybridization in amides lack knowledge. Only astute student C, who shuns both VSEPR and hybridization and searches for the three-dimensional structure beforehand, authentically constructs knowledge about amides.
Conclusion
Imagine learning about HAO and VSEPR for the first time - a daunting memory. If a bright student is interested enough to investigate further (such as reading this article), the result of advanced searches would bring conflicting and incompatible models, the use of multiple models for the same system, and even alternative models among classes of high school, introductory undergraduate, organic, inorganic and physical chemistry. Furthermore, HAO and VSEPR are found to be incompatible between themselves but are inexorably linked together.
In a second article, we provide a new framework to teach chemistry without the defunct models of HAO and VSEPR. Science advances only with the elimination of outdated concepts; chemical education needs a unified curriculum based on the latest experimental and computational results.

