











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las mitocondrias son organelos presentes en casi todas las células eucariotas, en las que ocupan hasta el 20% del volumen citoplasmático y son las responsables de proporcionar la energía necesaria para su sobrevivencia. Estos organelos evolucionaron a partir de alphaproteobacterias endosimbióticas dentro de una célula hospedadora derivada de arqueas del género Asgard (Roger et al., 2017). Con el tiempo, el ancestro bacteriano de la mitocondria se adaptó al contexto fisiológico del hospedero, lo que propició la remodelación de sus membranas, la aparición de las crestas mitocondriales y el desarrollo de procesos como la fusión y fisión mitocondrial (Pernas y Scorrano, 2016).
Las mitocondrias tienen forma ovoide con diámetros entre 0.5 y 1µm y se caracterizan por la presencia de dos membranas que forman cuatro compartimentos distintos: la matriz mitocondrial, la membrana interna, la membrana externa y el especio intermembrana (Pernas y Scorrano, 2016). La matriz es un espacio interno que contiene varias copias de DNA mitocondrial (DNAmt), así como enzimas necesarias para la oxidación de ácidos grasos, el ciclo de Krebs y otras que participan en la expresión de genes mitocondriales. La membrana interna es la que se pliega en numerosas ondulaciones llamadas crestas, las cuales desempeñan tres funciones principales: las reacciones de oxidación y reducción de la cadena de transporte de electrones, la producción de adenosín trifosfato (ATP, por sus siglas en inglés) mediante la fosforilación oxidativa y el transporte de metabolitos dentro y fuera de la matriz. Por su parte, la membrana externa se caracteriza por tener una gran cantidad de porinas permeables a moléculas de hasta 5,000 daltones y está involucrada en la interacción activa de la mitocondria con otros organelos celulares. Por último, el espacio intermembrana contiene varias enzimas que utilizan al ATP para fosforilar otras moléculas como la carnitina, implicada en el transporte ácidos grasos del citosol a la matriz mitocondrial (Giacomello et al., 2020)mitochondria are often considered the ‘powerhouses of the cell’. However, our understanding of the role of mitochondria in cell biology recently expanded when we recognized that they are key platforms for a plethora of cell signalling cascades. This functional versatility is tightly coupled to constant reshaping of the cellular mitochondrial network in a series of processes, collectively referred to as mitochondrial membrane dynamics and involving organelle fusion and fission (division.
Las mitocondrias son sumamente dinámicas, cambian constantemente de forma y localización dentro de la célula a través de procesos de fisión, fusión y transporte. Además, son los únicos organelos de las células animales que tienen su propio DNA. En humanos el DNAmt es circular y codifica para 13 subunidades de los complejos de la cadena de transporte de electrones; el resto de las subunidades son codificadas por el DNA nuclear, son sintetizadas en el citoplasma y transportadas hacia las mitocondrias (Raffaello y Rizzuto, 2011). De manera general, el NADH generado en el ciclo de Krebs y en la glucólisis es utilizado por las mitocondrias en la cadena de transporte de electrones y en la fosforilación oxidativa, para la producción de ATP, impulsada por un flujo de protones que genera un gradiente de cargas y pH (Mitchell, 1961), proceso conocido como potencial electroquímico. Durante este proceso, los electrones son transportados desde el NADH hasta el oxígeno por medio de complejos respiratorios y transportadores de electrones que están embebidos en la membrana interna de la mitocondria (Osellame et al., 2012).
Mitocondrias en las neuronas
El cerebro tiene una tasa metabólica alta: consume alrededor del 20% del oxígeno que se inspira en reposo y el 25% de la glucosa de todo el cuerpo, todo ello a pesar de que solo representa el 2% del peso corporal (Erecinska et al., 2004; Silver y Erecińska, 1998)energy (ATP. Dicha demanda metabólica se relaciona con el hecho de que las neuronas son células altamente diferenciadas que necesitan grandes cantidades de ATP para el mantenimiento de gradientes iónicos a través de las membranas celulares y para la neurotransmisión. Por ejemplo, el ATP generado por las mitocondrias es utilizado para dirigir el desmontaje de los complejos SNARE y el reciclamiento vesicular, incluyendo la escisión y el llenado de las vesículas sinápticas (Verstreken et al., 2005). Además, tras una estimulación eléctrica, las mitocondrias pueden amortiguar los niveles de calcio en la terminal presináptica (Rizzuto, 2001), lo que sugiere que pueden estar involucradas en la neurotransmisión (Levy et al., 2003).
Debido a su especialización morfológica y funcional, las neuronas no muestran una distribución uniforme de mitocondrias. Las áreas de alta demanda de ATP, como las terminales presinápticas, el cono de crecimiento axonal y los nodos de Ranvier, contienen más mitocondrias que otras partes de la neurona (Perkins et al., 2010; Smith y Gallo, 2018). Si bien, la biogénesis mitocondrial se puede producir en el axón, se piensa que la mayoría de las mitocondrias se generan en el soma, por lo que la neurona requiere de mecanismos especializados para su transporte. Dichos mecanismos están mediados principalmente por kinesinas y dineínas, además de proteínas acopladoras como miro, milton y sintafilina, que sirven como motores de desplazamiento y paro sobre el citoesqueleto de la neurona (Hollenbeck y Saxton, 2005). Se sabe que alrededor del 30% de las mitocondrias axonales son móviles (Sun et al., 2013) y se ha descrito que la dirección de su desplazamiento está relacionada con el potencial de membrana mitocondrial, de forma que las mitocondrias con un potencial de membrana alto se transportan en sentido anterógrado, mientras que las de menor potencial se desplazan hacia el soma neuronal.
Además de su transporte, las mitocondrias exhiben numerosos eventos de fusión y fisión, por lo que se les ha considerado como una red mitocondrial. La fusión proporciona un mecanismo por el cual la población de mitocondrias se mantiene homogénea y facilita la reorganización del contenido de la matriz de una mitocondria dañada con una sana, diluyendo las copias de ADNmt mutadas y el proteoma alterado (Chen et al., 2007; Tsakiri et al., 2019)the functional implication of the UPP in tissue homeodynamics at the whole organism level and its potential cross-talk with other proteostatic or mitostatic modules are not well understood. We show here that knock down (KD. Dicho evento está regulado por mitofusinas (Mfns) y OPA1, en donde las Mfns facilitan el anclaje de las membranas externas y las fusionan (Chen y Chan, 2009), mientras que OPA1, que se encuentra en la membrana interna, es responsable del remodelado y la unión de las crestas mitocondriales (Ishihara et al., 2006).
Por otro lado, la fisión mitocondrial ayuda a eliminar las mitocondrias dañadas de la célula (Kim et al., 2007) y participa en procesos como la formación de sinapsis y espinas dendríticas (Li et al., 2004). No obstante, el aumento excesivo en la fisión mitocondrial puede causar una incapacidad para generar ATP y aumentar la producción de especies reactivas de oxígeno (ERO) y con ello, incrementar la susceptibilidad de las células para experimentar apoptosis (Parone et al., 2008). La fisión está regulada principalmente por la proteína tipo dinamina 1 (Drp1) y el retículo endoplásmico (RE). Drp1 es una proteína citoplasmática que puede ser reclutada por varios acopladores mitocondriales como Fis1, MFF y Mid49, lo que genera que se formen complejos de Drp1 en forma de anillos que constriñen a la mitocondria, facilitando la escisión de su membrana doble (Smirnova et al., 2001). Recientemente se han definido dos tipos de fisión mitocondrial: La fisión mitocondrial por la zona media del organelo y la fisión periférica que ocurre en los extremos de las mitocondrias. Ambos tipos ocurren con frecuencia en células Cos-7 de monos, sin embargo, la fisión de la zona media ocurre en mitocondrias sanas mientras que la fisión periférica se lleva a cabo cuando una parte de la mitocondria presenta una disminución del potencial de membrana y un aumento de ERO (Kleele et al., 2021). De manera conjunta, el transporte, la fusión y la fisión mitocondrial engloban lo que se ha definido dinámica mitocondrial (Chen y Chan, 2009). Todo ello ha originado que surjan nuevos términos en el campo, por ejemplo, se ha acuñado el término mitostasis para hacer referencia a una forma especializada de homeostasis en donde el número y la calidad mitocondrial se mantienen a lo largo del tiempo en cada compartimento de la neurona (Misgeld y Schwarz, 2017).
Por otro lado, se han descrito nuevos procesos que evidencian más el papel dinámico de las mitocondrias. Recientemente, se reportó que existe una trasferencia mitocondrial entre neuronas y astrocitos luego de un evento de daño. Los astrocitos pueden suministrar mitocondrias funcionales a las neuronas dañadas para reestablecer sus propiedades energéticas (Hayakawa et al., 2016), en tanto que las neuronas pueden transferir sus mitocondrias dañadas a los astrocitos para ser degradadas (Davis et al., 2014). También se ha reportado la capacidad de liberar mitocondrias dañadas en vesículas denominadas exóforos de las neuronas de Caenorhabditis elegans (Melentijevic et al., 2017). De igual forma, se ha detectado que las mitocondrias fragmentadas liberadas por la microglía pueden desencadenar la respuesta astrocítica (Joshi et al., 2019), que los macrófagos pueden transferir mitocondrias a las neuronas sensoriales para resolver el dolor inflamatorio (Raoof et al., 2020), y que existe el tráfico de mitocondrias entre células troncales neuronales (Peruzzotti-Jametti et al., 2021). Lo anterior ha abierto la posibilidad de aprovechar la capacidad de las células de captar mitocondrias, para desarrollar estrategias de trasplante mitocondrial para contrarrestar el daño energético y funcional en patologías del sistema nervioso como ha ocurrido en otros modelos celulares (Emani y McCully, 2018; McCully et al., 2017; Gollihue et al., 2018). En este sentido, ya se ha empezado a evaluar el trasplante mitocondrial en el sistema nervioso mediante diversas estrategias de administración que parecen tener resultados prometedores (revisado en Espino De la Fuente-Muñoz y Arias, 2021).
Mitocondrias en la plasticidad neuronal
La plasticidad neuronal puede definirse como las adaptaciones estructurales y funcionales de los circuitos neuronales debidos al aprendizaje, la memoria, el ambiente y el daño cerebral y a la capacidad del cerebro para cambiar y adaptarse a un entorno que proporciona estímulos constantes (Citri y Malenka, 2008). Algunos ejemplos de plasticidad neuronal incluyen: la generación y el crecimiento de dendritas y axones, la neurogénesis, la formación de sinapsis y la potenciación a largo plazo (LTP, por sus siglas en inglés) (Cheng et al., 2010).
En este sentido, se ha encontrado evidencia que sugiere que las mitocondrias sinápticas son importantes en la transmisión sináptica mediante la regulación de calcio (Billups y Forsythe, 2002). Otros estudios han documentado cambios en la función mitocondrial durante la neurotransmisión, por ejemplo, durante la LTP la producción de energía y la expresión de genes mitocondriales aumenta (Abraham et al., 2002; Todorova y Blokland, 2016), en contraste con estudios farmacológicos que muestran que la inhibición de la actividad mitocondrial altera la neurotransmisión (Li et al., 2004). Estas observaciones son consistentes con el hecho de que el bloqueo de la fosforilación oxidativa conduce a un decremento de la LTP (Cunha et al., 1996). De la misma manera, utilizando carbonil-p(triflurometoxi) fenilhidrazona (FCCP), un desacoplante de la fosforilación oxidativa que colapsa el gradiente electroquímico de protones, o inhibiendo el complejo I mitocondrial con la toxina rotenona, se produce una menor liberación de vesículas en terminales nerviosas aisladas del hipocampo (Ivannikov et al., 2013), al igual que al inhibir la ATP sintasa con oligomicina (Verstreken et al., 2005). Aún más, se ha encontrado que la inducción de la LTP provoca una ráfaga rápida de fisión mitocondrial en las dendritas inducida por la activación de la calcio calmodulina quinasa II (CaMKII, por sus siglas en inglés) y la fosforilación de la proteína de fisión mitocondrial, Drp1 (Divakaruni et al., 2018).
Otro proceso involucrado en la plasticidad neuronal es la sinaptogénesis, durante la cual, el movimiento de las mitocondrias hacia las dendritas se correlaciona con la producción de energía y los cambios morfológicos de las espinas en desarrollo (Rangaraju et al., 2019; Rangaraju et al., 2019). La falta de proteínas de la dinámica mitocondrial como Drp1 y OPA1, disminuye el contenido de mitocondrias dendríticas y provoca una pérdida de contactos sinápticos, mientras que el aumento en el desplazamiento de las mitocondrias hacia las protuberancias dendríticas reestablece el número tanto de espinas como de sinapsis funcionales (Li et al., 2004). Aunado a ello, se ha demostrado que la regeneración axonal es deficiente cuando se inhibe el transporte mitocondrial, y en contraparte, la mejora del desplazamiento anterógrado de las mitocondrias incrementa su capacidad de regeneración (Zhou et al., 2016).
Otro proceso relevante en términos de plasticidad es la neurogénesis. Se sabe que la mayoría de las poblaciones de células troncales tienen un metabolismo glucolítico, con poca dependencia de la fosforilación oxidativa, fenómeno que se invierte cuando experimentan su diferenciación (Khacho et al., 2018). Pese a que las células troncales tienen poca dependencia de las mitocondrias para la generación de energía, existe evidencia de que desempeñan funciones en las células progenitoras neuronales. Así, durante la neurogénesis cortical embrionaria de ratones, las mitocondrias modifican su forma, siendo más alargadas dentro de las poblaciones celulares no comprometidas a un linaje celular, fragmentándose durante su diferenciación y elongándose una vez que las neuronas se han diferenciado (Khacho et al., 2016). Además, la inducción de la disfunción mitocondrial mediante la ablación del factor inductor de apoptosis, perjudica el desarrollo cortical embrionario presentándose anomalías en la auto renovación de células precursoras neuronales y en la diferenciación durante la neurogénesis (Khacho et al., 2017).
Implicación de las mitocondrias en procesos cognitivos
Como se ha descrito, las mitocondrias son importantes para los procesos de plasticidad en el cerebro, sin embargo, son pocos los estudios que abordan el papel de estos organelos en funciones mentales superiores como la cognición.
Uno de los primeros trabajos que se realizaron bajo este enfoque, determinó que un porcentaje elevado de heteroplasmia (presencia de ADN mitocondrial de distinto tipo en la misma célula), generaba un deterioro pronunciado en la memoria espacial a largo plazo en ratones, además de presentar deficiencias en la actividad del complejo IV, sugiriendo que la actividad mitocondrial es necesaria para la retención y consolidación de la memoria (Tanaka et al., 2008). Posteriormente, se demostró que la herencia de altos niveles de heteroplasmia produce déficits metabólicos y cognitivos (Sharpley et al., 2012). Por su parte, el análisis en sinaptosomas (terminales presinápticas) aislados de monos, mostró una relación entre la morfología mitocondrial, la función cognitiva y el envejecimiento, ya que los sinaptosomas de monos viejos tienen numerosas mitocondrias en forma de rosquilla, lo que es indicativo de estrés mitocondrial (Ahmad et al., 2013; Hara et al., 2014). Esta morfología se correlacionó con una memoria de trabajo reducida, pérdida de sinapsis y un número disminuido de vesículas sinápticas, sugiriendo que la morfología mitocondrial, y por lo tanto su funcionalidad, influye en el rendimiento cognitivo durante el envejecimiento (Hara et al., 2014).
Recientemente, se ha descrito que algunos receptores de la membrana mitocondrial también tienen implicaciones en la memoria, en particular la activación de los receptores a cannabinoides tipo 1 de la mitocondria pueden participar en la transmisión sináptica y la formación de la memoria a través de la modulación de la señalización de proteínas G intramitocondriales (Hebert-Chatelain et al., 2016). Otros modelos experimentales han demostrado la participación de las mitocondrias en el deterioro de la memoria inducido por escopolamina, un compuesto que interfiere con la transmisión colinérgica en el sistema nervioso central y provoca deterioro cognitivo (Wong-Guerra et al., 2017), y en los efectos de la administración crónica de tramadol, un analgésico de tipo opioide que causa disfunción mitocondrial y se correlaciona con el afectación de la memoria espacial en ratas, efectos que son mitigados con ejercicio físico (Mehdizadeh et al., 2017).
Otros eventos que se han visto implicados en procesos de memoria y aprendizaje son la dinámica mitocondrial y la autofagia, ya que, la inhibición de Drp1 disminuye la fragmentación mitocondrial excesiva, la deposición del péptido amiloide-β (Aβ) y mejora la conducta de ratones en modelos de la Enfermedad de Alzheimer (EA) (Baek et al., 2017). Por su parte, la quinasa 1 inducida por PTEN (PINK1, por sus siglas en inglés) implicada en la regulación de la mitofagia, se ha relacionado con efectos positivos en procesos de memoria y aprendizaje, ya que su eliminación conlleva a alteraciones en la memoria espacial, mientras que la restauración de su función promueve la eliminación de mitocondrias dañadas (Du et al., 2017) y previene y revierte el deterioro cognitivo en modelos animales de la EA (Fang et al., 2019).
Los ejemplos anteriores muestran la creciente evidencia del papel que tienen las mitocondrias en el sistema nervioso, particularmente en los procesos de plasticidad neuronal.
Disfunción mitocondrial en la Enfermedad de Alzheimer
La EA es una enfermedad neurodegenerativa que se caracteriza por la pérdida progresiva de la memoria, disfunción cognitiva, alteraciones en el comportamiento, delirios y el detrimento de la convivencia social, así como una disminución progresiva del lenguaje (Selkoe, 2001). Hasta el momento se sabe que la EA es una enfermedad multifactorial que tiene a la edad como el principal factor de riesgo. Las características histopatológicas asociadas a la enfermedad son las placas amiloideas formadas por agregados de Aβ, las marañas neurofibrilares de la proteína tau hiperfosforilada y una extensa pérdida neuronal (Long y Holtzman, 2019; Querfurth y LaFerla, 2010). Además, se ha planteado que la EA es el resultado de una falla sináptica (Selkoe, 2002), ya que los pacientes presentan una reducción del 25% al 35% en la densidad numérica de las sinapsis de manera temprana (Davies et al., 1987), así como una reducción en los niveles de proteínas sinápticas como sinaptofisina, sinaptotagmina y GAP43 (Masliah et al., 2001).
A pesar de que ciertos factores que subyacen a la enfermedad se conocen bien, actualmente existen varias líneas de investigación que sugieren que las mitocondrias están implicadas de manera importante en el desarrollo y la progresión de la EA. Se sabe que en pacientes con EA, el número de mitocondrias y de crestas mitocondriales disminuye (Hirai et al., 2001) (Figura 1), se presenta un aumento en el contenido de las proteínas que regulan la fisión mitocondrial, así como una disminución en los niveles de las proteínas que regulan la fusión (Manczak et al., 2011; Wang et al., 2009). Por otro lado, en cerebros de pacientes, la expresión de genes relacionados con la cadena de transporte de electrones y el ciclo de Krebs se ve afectada (Brooks et al., 2007; Liang et al., 2008) mientras que la actividad de algunas enzimas involucradas en estos procesos se ve disminuida (Bosetti et al., 2002; Butterworth y Besnard, 1990; Mastrogiacomo et al., 1996). Un análisis bioinformático del hipocampo de pacientes identificó a la fosforilación oxidativa como una de las vías más importantes implicadas en la EA (Zhang et al., 2015), y el análisis de enriquecimiento de genes demostró que existe una regulación a la baja de la fosforilación oxidativa y una alteración de las vías de importación y proteostasis mitocondrial (Sorrentino et al., 2017). Estos resultados son respaldados por la disminución de los niveles de Tom20 y Tom70 (Chai et al., 2017) y la disminución de la actividad enzimática de la proteasa PreP, encargada de escindir a Aβ dentro de la mitocondria (Alikhani et al., 2011; Mossmann et al., 2014).
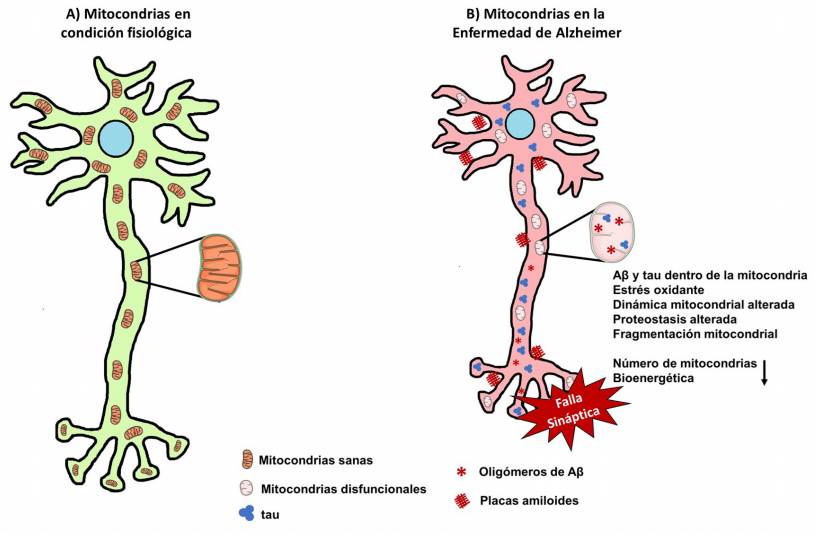
Figura 1 Alteraciones mitocondriales durante la Enfermedad de Alzheimer. A) Las mitocondrias en condiciones fisiológicas son dinámicas y migran lo largo del soma, dendritas y axón neuronales. B) En la Enfermedad de Alzheimer existen alteraciones en la dinámica y bioenergética mitocondrial, así como acumulación de Aβ y tau. La disfunción de las mitocondrias juega un papel importante en el daño sináptico y la muerte neuronal.
En relación con lo anterior, se han realizado estudios donde se presentan alteraciones mitocondriales conforme se desarrollan los marcadores histopatológicos de la enfermedad, la mayoría de ellos enfocados en entender el papel que tiene el péptido Aβ en la disfunción mitocondrial. Por ejemplo, cuando neuronas hipocampales de rata son tratadas con Aβ, la densidad de las neuritas y la longitud de las mitocondrias disminuyen (Wang et al., 2009). De manera similar, en células de neuroblastoma que sobreexpresan mutaciones humanas de la proteína precursora de Aβ (APP), se observa un incremento en la fragmentación mitocondrial y un cambio en la distribución de las mitocondrias hacía la zona perinuclear (Wang et al., 2008). Por su parte, en ratones mAPP se presenta una disminución en la actividad enzimática del complejo IV y una desregulación en el transporte axonal (Du et al., 2010).
También se ha documentado la interacción de Drp1 con el Aβ en pacientes con la EA y en ratones que sobreexpresan APP, lo que sugiriere que esta interacción puede ser un factor relevante para la fragmentación anormal de las mitocondrias y su disfunción mediada por el Aβ (Manczak et al., 2011). Además, se ha observado que existe una disfunción bioenergética en mitocondrias aisladas de cerebros en modelos de la enfermedad como es el ratón 3xTg-AD (Yao et al., 2009) y un desbalance en la dinámica y en el potencial de membrana mitocondrial en ratones 5xFAD (Wang et al., 2016), similar a lo reportado en ratas transgénicas que sobre expresan APP humana mutada (Adami et al., 2017). Recientemente, hemos descrito que existe una disminución metabólica y bioenergética en sinaptosomas aislados del hipocampo, la corteza cerebral y el cerebelo de ratones 3xTg-AD, la cual se asoció con la fragmentación mitocondrial mediada por Drp1, así como la acumulación de Aβ y tau dentro de las mitocondrias sinápticas (Espino De la Fuente-Muñoz et al., 2020) (Figura 1).
En relación con lo anterior, varios trabajos han encontrado la presencia de Aβ en la matriz y en las crestas mitocondriales de neuronas (Cenini et al., 2016; Du et al., 2010; Espino De la Fuente-Muñoz et al., 2020; Hansson Petersen et al., 2008; Manczak et al., 2006). En este sentido, cada vez más se tiene un mayor conocimiento de cómo es que el péptido se internaliza en estos organelos. Por un lado, se ha identificado una forma de APP mitocondrial, no obstante, se ha demostrado que las mitocondrias no poseen la maquinaria enzimática para producir Aβ (Mamada et al., 2017), ya que el APP mitocondrial se escinde en el espacio intermembrana por la proteasa Omi, generando un fragmento diferente del Aβ (Park et al., 2006), aunque el APP mitocondrial puede acumularse en los canales de importación impidiendo el tráfico de proteínas codificadas en el núcleo (Devi et al., 2006). Además, se sabe que el Aβ es transportado hacia el interior de la mitocondria por medio de la translocasa de la membrana externa (TOM, por sus siglas en inglés) y que su acumulación bloquea la entrada de proteínas citoplasmáticas a la matriz mitocondrial (Hansson Petersen et al., 2008; Manczak et al., 2006). Por otro lado, el péptido Aβ se produce de manera abundante en las membranas asociadas a mitocondrias (MAMs), que son sitios de contacto y comunicación entre las mitocondrias y el RE y que participan en la fisión mitocondrial y la homeostasis de calcio (Fonseca et al., 2015; Raturi y Simmen, 2013). Las MAMs se encuentran incrementadas en fibroblastos de pacientes con la EA y en modelos transgénicos, lo que sugiere que el aumento de estos contactos podría favorecer la producción de Aβ cerca del sistema de importación mitocondrial (Area-Gomez y Schon, 2016).
Una vez que el Aβ ha sido importado a la mitocondria causa diferentes alteraciones, siendo el complejo IV el más afectado tanto en modelos que expresan APP humana como en mitocondrias cerebrales de pacientes con la EA (Devi et al., 2006; Rönnbäck et al., 2015). Existe evidencia de que el complejo IV (Atamna y Boyle, 2006; Crouch et al., 2006; Hernandez-Zimbron et al., 2012) y la ATPasa (Beck et al., 2016), son inhibidos de manera directa por la unión del Aβ.
Además del Aβ, otra de las proteínas involucrada en la EA, la proteína tau, también ha sido asociada con alteraciones mitocondriales. Usando diferentes modelos se ha demostrado que la sobreexpresión y la hiperfosforilación anormal de tau en los sitios AT8, inhiben el transporte de las mitocondrias a lo largo de los axones de neuronas corticales, contribuyendo a la degeneración axonal (Cabezas-Opazo et al., 2015; Shahpasand et al., 2012). Por otro lado, también se sabe de la interacción anormal entre tau hiperfosforilada y Drp1, lo que tiene como consecuencia una fisión mitocondrial excesiva que conduce a la degeneración de las sinapsis en los ratones 3xTg-AD (Manczak y Reddy, 2012). En un estudio sobre los efectos de expresar diferentes formas truncadas de tau en neuronas, se encontró una reducción de los niveles de OPA1, lo que podría tener implicaciones en la remodelación de las crestas mitocondriales y con ello en la eficiencia de la cadena de transporte de electrones (Pérez et al., 2018).
Tanto en cultivos celulares como en ratones transgénicos, la sobreexpresión de tau inhibe la función mitocondrial al disminuir la actividad de los complejos respiratorios y las enzimas antioxidantes (Cheng y Bai, 2018). Así mismo, se ha encontrado que tau se puede unir a la membrana externa de la mitocondria y afectar el potencial de membrana mitocondrial (Atlante et al., 2008). La interacción anormal de tau fosforilada con la proteína del canal aniónico dependiente de voltaje (VDAC1) se ha observado en pacientes y en el modelo 3xTg-AD, lo que altera la apertura y el cierre de los poros mitocondriales provocando disfunción mitocondrial neuronal (Manczak y Reddy, 2012). Por último, estudios proteómicos y funcionales en ratones que sobre expresan la mutante P301 de tau, demostraron que estos ratones desarrollaron disminución en la actividad de los complejos mitocondriales, incrementos en la producción de ERO y despolarización mitocondrial (David et al., 2005; Rhein et al., 2009).
Conclusiones
Durante varios años se ha documentado la relevancia que tienen las mitocondrias como las centrales energéticas de las células eucariotas y existe gran evidencia de la importancia que tienen estos organelos en células altamente especializadas como las neuronas y el papel de su disfunción en procesos que subyacen a la plasticidad neuronal. En contraste, pocos son los estudios que se han enfocado en analizar la relación que existe entre los procesos cognitivos y las mitocondrias desde una perspectiva fisiopatológica. Sin embargo, es un campo que ha ido creciendo y que está contribuyendo a entender los mecanismos que controlan la memoria y el aprendizaje y sus alteraciones en el envejecimiento y en las enfermedades neurodegenerativas como la EA, principalmente sobre la afectación bioenergética y las alteraciones de la dinámica mitocondrial.
Se ha descrito que en la EA ocurre daño mitocondrial, pero aún se debate si esta alteración precede al desarrollo de la patología o es un evento que se desencadena una vez que se presenta el padecimiento. Por un lado, proteínas asociadas con la EA como el Aβ y la tau, pueden inducir efectos nocivos en la mitocondria y podrían iniciar una cascada de daño secundaria. Por otro lado, el daño mitocondrial podría presentarse en primera instancia, de manera temprana, de tal forma que los déficits bioenergéticos afecten directamente las funciones neuronales y las lleven a la muerte. En cualquiera de los dos casos, es claro que las alteraciones mitocondriales contribuyen al daño neuronal, por lo que, abordar la disfunción neuronal de la EA desde una perspectiva terapéutica enfocada en la restauración de la función mitocondrial, es atractiva y empieza a mostrar resultados interesantes (revisado en Espino De la Fuente-Muñoz y Arias, 2021).

