











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Los diagramas de fases de presión (P) versus temperatura (T) de un componente, suelen presentarse por primera vez a los alumnos en la mayoría de los cursos universitarios de química básica. En muchos libros de texto de química o fisicoquímica, se discuten los diagramas de fases representativos de aquellas sustancias donde -salvo en condiciones extremas de P- el número de fases se limita a tres (los estados de agregación sólido, líquido y gaseoso). Usualmente, sólo se incluyen los diagramas para H2O y CO2, y limitados, especialmente en el primer caso, a presiones apenas superiores a la del punto crítico (218 atmósferas). En un nivel de complejidad un poco mayor, y para mostrar un ejemplo de diagrama de fases con más de una fase sólida (es decir, donde el diagrama de fases es algo más que un diagrama de estados de agregación), es muy común que los libros de texto elijan el diagrama de fases del azufre. Si bien, el polimorfismo (alotropía) del azufre es muy complejo (Cotton y Wilkinson, 1975; MacKay, MacKay y Henderson, 2002; Meyer, 1976), a presiones por debajo de las 2000 atmósferas, sólo existen dos fases sólidas estables. Esto lo convierte en un ejemplo más complejo, pero aún accesible para la comprensión de los recién iniciados en el tema.
En la mayoría de los casos los diagramas son esquemáticos y fuera de escala para poder abarcar un rango suficiente de presiones y temperaturas. Además, es usual que se exageren algunos detalles o se modifiquen escalas en intervalos dentro de un mismo diagrama, en función de exaltar determinadas características de las sustancias y/o de sus cambios de estado. Los diagramas, como todas las representaciones gráficas, son extremadamente poderosos para descubrir rápidamente las relaciones entre variables. Esto permite la interpretación visual de los datos muy rápidamente. No obstante, esas ventajas de una representación a mano alzada pueden convertirse en una fuente de conceptos o de interpretaciones erróneas, si se cometen ciertos descuidos gráficos. El reconocimiento de las distorsiones sustentadas por razones didácticas o prácticas, así como la prevención de aquellas que no están justificadas en comportamientos cualitativos reales, permiten evitar dichas concepciones equívocas. De esta manera, además, se evitan inconvenientes futuros durante el uso de los diagramas de fases.
En este trabajo discutiremos algunas concepciones equívocas que son inducidas a partir de errores de representación esquemática en el diagrama de fases del azufre, y que son usuales no sólo en libros de química y fisicoquímica sino también en algunos artículos científicos. También se presenta un tratamiento, a partir de datos reales, para la construcción del diagrama de fases a escala.
El diagrama de fases del azufre
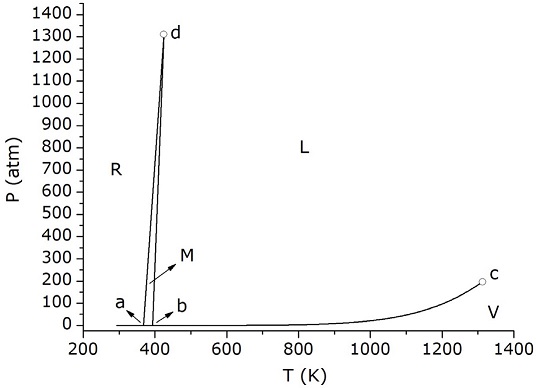
A presiones no demasiado elevadas, las únicas fases sólidas estables para el azufre son la alfa (conocida como fase rómbica, R) y la beta (conocida como fase monoclínica, M). En la Figura 1 se presenta un diagrama de fases del azufre (fuera de escala). Las transformaciones en el azufre (Meyer, 1976) son lentas (debido a la baja conductividad térmica) y durante ellas, pueden darse cambios en la composición molecular (Sn, con n = 8 para azufre rómbico, a n < 8 para otros estados). Por ello, no hay acuerdo exacto en todos los valores de las presiones y temperaturas de los puntos triples a, b y d (las mediciones de diferentes autores pueden diferir más allá del error experimental). Los valores consignados en la Figura 1 corresponden a los reportados por Ferreira y Lobo, (2011) y, al igual que estos autores, aquí supondremos que todas las fases están compuestas por moléculas S8.
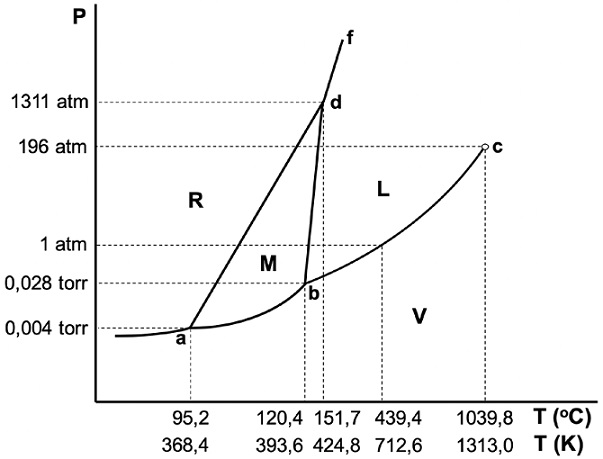
Figura 1 Diagrama de fases del azufre (fuera de escala). Las zonas monofásicas son: sólido rómbico (R), sólido monoclínico (M), líquido (L) y vapor (V). El punto c es el punto crítico.
La Figura 1, si bien está fuera de escala, no tiene características que, desde el punto de vista cualitativo, muestre algún comportamiento incorrecto en el sistema. Veremos, a continuación, los errores de representación frecuentes.
El equilibrio monoclínico-líquido (M-L)
Un error muy común es el que se observa en la Figura 2. En esa figura la curva b-d que corresponde al equilibrio entre la fase monoclínica y la fase líquida (fusión del azufre monoclínico) tiene una pendiente negativa. Este error probablemente se origina de manera involuntaria al pretender encerrar la fase monoclínica en una zona, limitada, además, por los equilibrios de transición entre azufre rómbico y azufre monoclínico y de sublimación de la fase monoclínica. No obstante, la pendiente real no es negativa sino positiva. La explicación está al alcance de los contenidos de una química general. La pendiente, dP/dT, de cada curva de equilibrio entre dos fases X e Y está dada por la ecuación de Clapeyron (Atkins y de Paula, 2008, p. 126), es decir
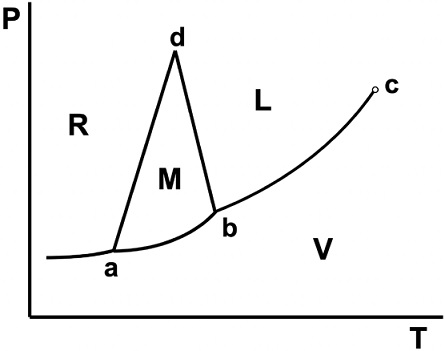
Figura 2 Un clásico error: la pendiente negativa para la curva b-d (equilibrio monoclínico-líquido).
donde ∆HX-Y es la entalpía molar (calor latente) para el cambio de fase, T es la temperatura absoluta (en K), mientras que vX y vY son los volúmenes molares de las fases en equilibrio. Para evaluar la pendiente de las curvas R-M y M-L, son necesarios los datos de la ecuación 1 para los procesos RM y ML. Para la temperatura correspondiente al punto d, Las entalpías (calculadas a presión de 1 atm), supuestas muy poco dependientes de la presión (y, por ende, válidas a la presión correspondiente al punto d), son (Ferreira y Lobo, 2011), ∆HRM = 3034 J/mol y ∆HML = 16149 J/mol. Para los volúmenes molares (en el punto d), se tiene (Ferreira y Lobo, 2011), vR = 127,272 cm3/mol, vM = 130,652 cm3/mol y vL = 139,071 cm3/mol. Por lo tanto, empleando la ecuación 1, se puede establecer que ambas curvas poseen pendiente positiva, siendo la pendiente de la curva R-M (2,11 J/K cm3 = 20,8 atm/K) menor que la de la curva M-L (4,52 J/K cm3 = 44,6 atm/K). Esta diferencia en pendiente explica por qué finalmente ambas curvas se encuentran en el punto d. Debido a que las pendientes de las curvas de equilibrio entre fases condensadas serán prácticamente constantes, los valores calculados en el punto d se suponen válidos para el rango completo de presión y temperatura. Por ende, las pendientes de las curvas R-M y M-L también pueden estimarse suponiendo comportamiento lineal entre los valores consignados en la Figura 1. Considerando una presión igual a 0 para los puntos a y b, Los valores que se obtienen son 23,4 atm/K y 42,3 atm/K para los equilibrios R-M y M-L, respectivamente. Estos valores, semejantes a los calculados previamente con la ecuación de Clapeyron, indican que ambas curvas son muy empinadas (típico de equilibrios entre fases condensadas). Esto provoca que no sea posible visualizarlas de ese modo en un diagrama fuera de escala. De dichos valores para las pendientes se puede calcular que los aumentos de temperatura provocados por cada incremento de la presión en una atmósfera son muy pequeños, es decir 0,04 oC/ atm (R-M) y 0,02 oC/atm (M-L). Pero incluso corrigiendo el error de la Figura 2, la distorsión que se produce en un gráfico como el de la Figura 1, puede aún inducir a suponer un cambio significativo en las temperaturas de transformación RM y ML a la presión de 1 atm, respecto de los puntos triples a y b, respectivamente. En realidad, son prácticamente los mismos valores dentro de las centésimas de oC.
Una posibilidad para evitar esta distorsión provocada por un diagrama fuera de escala es utilizar un diagrama con representación logarítmica de la presión como esquemáticamente muestra la Figura 3. En ella queda claro que las curvas de equilibrio entre fases condensadas (R-M y M-L) son prácticamente verticales en la zona de presiones bajas del diagrama.
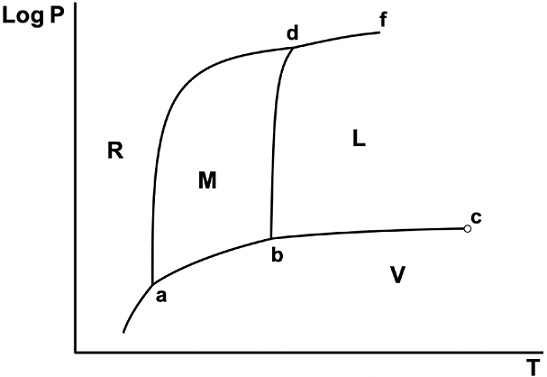
Aunque el gráfico en escala logarítmica (estrictamente, semilogarítmica ya que se aplica logaritmos sólo a la variable presión) permite visualizar mejor los equilibrios entre fases condensadas, resulta más difícil de interpretar. La utilización de una escala logarítmica hace que multipliquemos por una cantidad fija (en el caso del logaritmo decimal, por diez) el valor unitario de la magnitud, en lugar de sumárselo como se hace en una escala lineal. La escala logarítmica tiene un crecimiento más lento ya que un determinado intervalo corresponde a un cambio mucho mayor de la variable. Este aspecto también provoca una distorsión en la interpretación del comportamiento del sistema, haciendo que cuanto más nos alejamos del punto de partida, más plana sea la curva. Así, en el diagrama de fases de la Figura 3, las rectas que limitan la zona de estabilidad de la fase monoclínica se vuelven curvadas en las cercanías del punto triple d, y los equilibrios con el vapor, se aplanan y/o invierten su curvatura (respecto de un gráfico en escala lineal). De aquí que la curva de equilibrio R-L (la zona del gráfico más allá del punto d) puede sugerir grandes cambios de las temperaturas de equilibrio para variaciones relativamente pequeñas de la presión, lo que cual es una imagen incorrecta producto de la aplicación de la función logaritmo.
Los puntos triples como puntos de inflexión
En las Figuras 1-3 se observan los puntos triples a y b como puntos de una significativa inflexión entre las curvas de equilibrio. Sin dudas, esta representación tiene una motivación didáctica que resalta la ubicación de los puntos triples y que señala que estos puntos muestran cierto cambio en el comportamiento de la presión de vapor con la temperatura. Las curvas de equilibrio entre una fase condensada y vapor (R-V, M-V y L-V) que se cruzan en estos puntos triples también responden a la ecuación de Clapeyron (ecuación 1). Una aproximación válida para esos tres equilibrios es considerar que la variación de volumen coincide con el volumen molar del vapor, ya que éste será (excepto a temperaturas cercanas a la del punto crítico c) significativamente superior al de la fase condensada correspondiente1. Al utilizar esta aproximación, resulta evidente que las pendientes de las curvas R-V y M-V en a y las pendientes de las curvas M-V y L-V en b serán sólo proporcionales a las variaciones de entalpía de los procesos correspondientes a las curvas que se encuentran en cada punto (ya que la temperatura y el volumen del vapor coincidirán). Debido a que las variaciones de entalpía siguen el orden decreciente ∆HRV > ∆HMV > ∆HLv, se espera que, para una representación en escala lineal, la pendiente R-V sea mayor a la M-V en a y la M-V mayor que L-V en b, tal cual sugieren las Figuras 1 y 2.
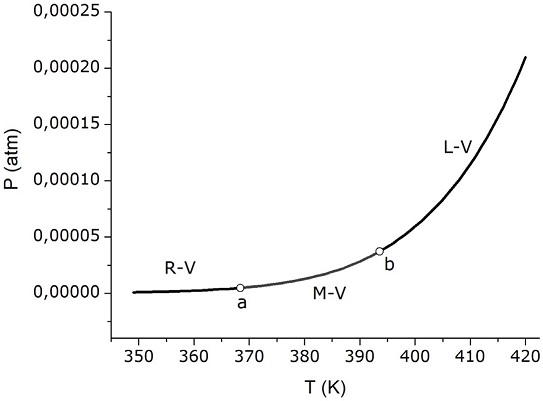
No obstante, cabe preguntarse si este comportamiento cualitativamente justificado por la ecuación de Clapeyron es una representación adecuada de los datos experimentales, o también podría tratarse de una distorsión del esquema con fines didácticos. Para dilucidar esta cuestión, representaremos los datos experimentales, a escala real, para las curvas de equilibrio entre las fases estables (es decir, en el tramo que incluye a los puntos triples a y b). Utilizaremos para ello ecuaciones que ajustan a los resultados experimentales y que fueron reportadas previamente. Para las curvas de sublimación de las fases R y M, los ajustes de Neumann (1934), adaptados a la función exponencial y a unidades de presión en atm, son los siguientes (T en K):
Equilibrio R-V:
Equilibrio M-V:
Y para el equilibrio L-V, el ajuste de Ferreira y Lobo (2011) es:
donde, Pc es la presión crítica (196 atm), Tc es la temperatura crítica (1313 K), τ se calcula como 1 - (T/Tc) y k es una constante de valor igual a -5,47×10-6 atm (esta última es una pequeña corrección aplicada a la fórmula original para lograr la continuidad en el punto b). La Figura 4 muestra la gráfica resultante. Como se observa, dentro de la precisión propia de los ajustes, no se aprecian las inflexiones en los puntos triples. Esto coincide con lo que se ha reportado para otros diagramas de fases (Hawkes, 1999). Cuando se realiza un análisis cuantitativo, se encuentra que el cambio relativo de pendientes es muy pequeño. En el caso del punto triple a, y a partir de los valores de ∆HRV y ∆HMV, puede comprobarse que la pendiente inicial M-V es apenas 1/1000 menor que la pendiente final R-V. Por ende, la diferencia es imperceptible en cualquier escala lineal que se utilice. Si bien, para un diagrama fuera de escala, la exageración de las inflexiones está de algún modo justificada para mostrar inequívocamente la posición de los puntos triples, resulta importante la discusión complementaria sobre el verdadero aspecto del diagrama de fases en torno a esos puntos. En función de ello, en la Figura 5 se muestra el diagrama de fases a escala, construido a partir de los ajustes mostrados en la Figura 4 y a los cuales se les añadieron las curvas R-M y M-L. Respecto de Las curvas R-M, M-L, dado el rango alto de presiones, los ajustes parabólicos (Ferreira y Lobo, 2011) no pasan exactamente por los puntos triples a, b y d. De modo que un ajuste lineal a base de dichos puntos fijos resulta una alternativa adecuada. Dichas ecuaciones son (T en K):
El equilibrio rómbico-líquido (R-L)
Otro error muy frecuente se encuentra en la descripción del comportamiento del sistema para presiones y temperaturas superiores a las del punto d, que es un tercer punto triple en el diagrama. Este error no tiene, en principio, ninguna justificación didáctica y tampoco razones de escala que lo provoquen y podría asociarse a simples descuidos. Suele manifestarse en dos variantes distintas, tal cual muestra la Figura 6: la línea correspondiente a la coexistencia de la fase R y la fase L (curva d-f o curva R-L), se considera prolongación de la curva R-M (caso A), o como prolongación de la curva M-L (caso B). Ninguna de las dos extrapolaciones es correcta, y la respuesta a cómo es el comportamiento correcto de la curva de equilibrio R-L, se obtiene fácilmente, mediante un razonamiento análogo al aplicado a la curva M-L. Con los valores ya empleados de entalpías de transformación para el punto d, podemos calcular la entalpía de fusión de la fase R, es decir: ∆HR→L = ∆HR→M + ∆HM→L = 19183 J/mol (Ferreira y Lobo, 2011). Aquí también vale la aproximación de comportamiento lineal (para equilibrios de fases condensadas), de manera que la ecuación 1 permite calcular la pendiente de dicha recta en el punto d (empleando nuevamente los volúmenes de fase indicados más arriba), lo cual da 3,82 J/K cm3, (37,7 atm/K). Este es un valor intermedio entre las pendientes de las curvas R-M y M-L en el mismo punto. Por lo tanto, en el diagrama, el equilibrio R-L se debe señalar tal como se indica esquemáticamente en la Figura 1 (segmento d-f). Ese mismo comportamiento se intenta reflejar en el diagrama semilogarítmico de la Figura 2, lo que generalmente se hace de manera ambigua en algunos libros de texto y artículos. Más allá de su pequeña diferencia visual con las alternativas erróneas, esta corrección está dirigida a distinguir la curva R-L como una curva independiente de los equilibrios en los que está involucrada la fase M y no como una continuación de alguno de ellos.
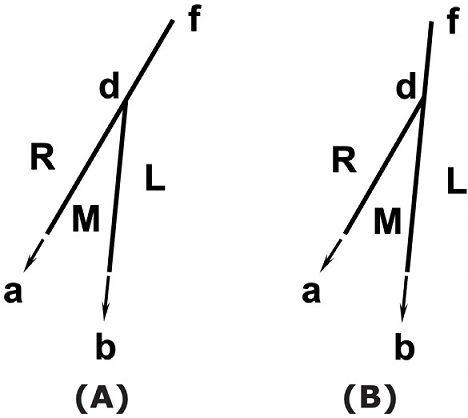
Un cuarto punto triple
Una de las consecuencias interesantes de la mencionada lentitud de los cambios en este sistema, es sobre la transformación RM, la cual requiere de tiempos del orden de los días para alcanzar el equilibrio. De este modo, un calentamiento normal de la fase R permite mantenerla (sin que se transforme en M) a temperaturas más allá de las correspondientes a la curva R-M. Ese estado metaestable se describe como un sobrecalentamiento de la fase R que es semejante al que sufre el agua líquida cuando, por ejemplo, a una presión de 1 atm, es sobrecalentada por encima de 100 oC. En el caso del azufre, la fase R puede incluso fundir sin que haya aparecido la fase estable M. Este comportamiento de fusión metaestable de R resulta estar descrito por la extrapolación del equilibrio estable R-L (comportamiento lineal d-f en Figuras 1 y 3) hacia bajas presiones dentro del área triangular abd, que es la zona de la fase M estable. De modo que es previsible la existencia de un punto triple e para el equilibrio metaestable de la fase R con las fases L y V, que será la intersección de la recta del equilibrio metaestable R-L con las curvas de los equilibrios metaestables R-V y L-V, tal como se señala esquemáticamente en la Figura 7. Debido a la imperceptible inflexión en los puntos triples a y b (ver Figura 4 y el texto relacionado), no es posible determinar las coordenadas del punto e mediante extrapolación, gráfica o analítica, de los comportamientos R-V y L-V. A partir de datos experimentales del equilibrio R-L (Tammann, 1903), en el rango 394 K < T < 439 K que incluye ambas zonas, la estable y la metaestable, es posible realizar un ajuste lineal (coeficiente de correlación 0,9997) correspondiente a la recta e-f que queda expresado por la ecuación (T en K)
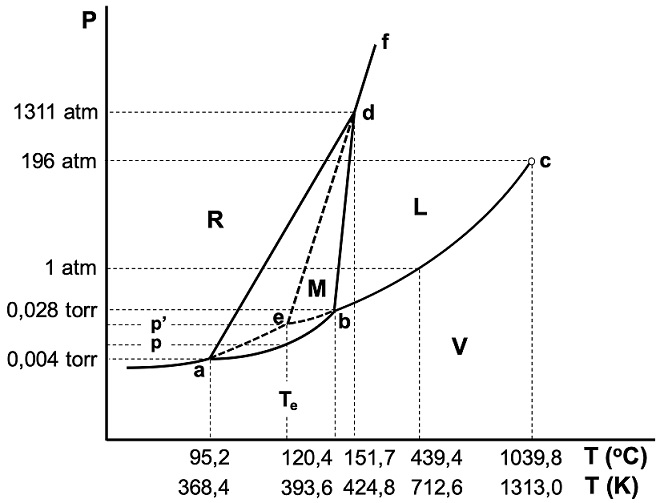
La pendiente (37,4 atm/K) que surge de esta ecuación es similar a la pendiente calculada más arriba, a partir de la ecuación 1, para el equilibrio R-L estable. La expresión 7 permite estimar la temperatura correspondiente al punto triple metaestable, Te, si se extrapola a P = 0 (lo cual equivale como aproximación a la presión del punto e), es decir Te = 387 K. Para averiguar la presión correspondiente, se efectuará un cálculo termodinámico.
A la temperatura Te, la fase R es menos estable que la fase M, por lo que se espera que su presión de vapor (p’) sea mayor a la presión de vapor de la fase M (p) a la misma temperatura (ver Figura 7). Y, tal como es posible inferir a partir de la Figura 4, ambas presiones de vapor tienen que ser muy cercanas. La dependencia con la presión de la energía libre de Gibbs de la fase vapor, V, (considerando comportamiento ideal), está dada por (Atkins y de Paula, 2008, p. 109)
donde Go(T) es la energía libre estándar de Gibbs (P = 1 atm), a la temperatura T. Para sólidos, el efecto de la presión en el rango de 1 atm es despreciable. La Figura 8 muestra de manera esquemática las variaciones de la energía libre de Gibbs con la presión para las fases sólidas, R, M y para el vapor (para una fase, la pendiente de las curvas, [∂G/∂P]T es el volumen de la fase, v, siendo en este caso, vV >> vM > vR). El equilibrio metaestable M-V a la temperatura Te (Figura 7) involucra el cruce de GV(Te,p) y GM(Te,p). El equilibrio metaestable R-L-V en el punto triple e, significa el cruce GV(Te,p’) y GR(Te,p’), donde p’ es la presión de vapor de la fase R a la temperatura Te. De este modo:
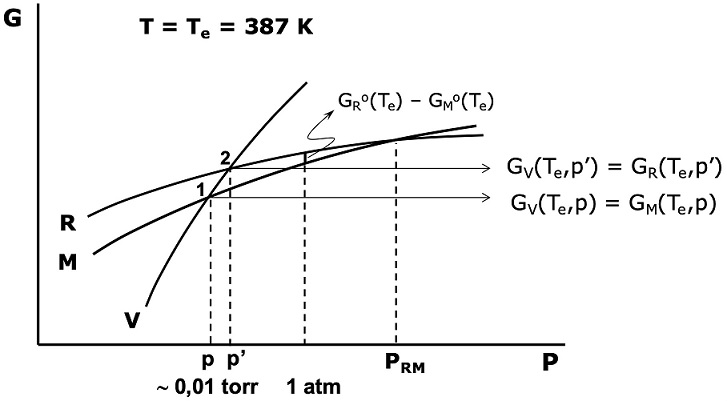
Figura 8 Variación de Energía libre de Gibbs con la presión para las fases solidas (R y M) y el vapor (V), a la temperatura Te.
Empleando la ecuación 8, el primer miembro de la ecuación 9 se puede escribir como RT ln(p’/p). El segundo miembro, debido a la mencionada independencia de la función G con la presión, puede considerarse igual a GR o(Te) - GM o(Te) (ver Figura 8) y equivale a -∆f,MGo(Te) que es la energía libre estándar de Gibbs para la formación de la fase M (respecto de la R, que es el estado estándar) a la temperatura Te y cambiada de signo. Entonces, la ecuación 9 se transforma en
Considerando que Te es 387 K, a partir de datos de tablas (Chase, 1998) para la energía libre de formación de la fase M, se puede extrapolar ∆f,MGo(Te) = - 0,166 kJ/mol S8 (a una temperatura mayor que 368,4 K, la fase M es la fase sólida estable). Luego, de la ecuación 10, se calcula p’/p = 1,05. Finalmente, la ecuación 3 permite obtener el valor p(Te) resultando 2,24×10-5 atm (0,017 torr). De este modo, se puede obtener el valor p’ = 1,05 p = 2,36×10-5 atm (0,018 torr). La pequeña diferencia entre p y p’ (10-3 torr) hace notar un detalle más en el diagrama de fases fuera de escala, sobre el cual debe tenerse cierta precaución. En cualquier diagrama en una escala no distorsionada en el que se pretenda ver los cuatro puntos triples y el punto crítico, no existen chances de distinguir la posición del punto e respecto de la curva de equilibrio M-V. Las Figuras 9a (con escala lineal) y 9b (con escala semilogarítmica) muestran precisamente la posición en la que quedaría e en un diagrama no distorsionado. En la escala lineal, el punto e parece erróneamente ubicado sobre la curva M-V. Esta dificultad se elimina al utilizar la escala logarítmica; una ampliación de la zona en esta última escala (Figura 9c) permite visualizar que el punto triple metaestable está apenas desplazado hacia arriba respecto del punto estable de equilibrio de fases (M-V) correspondiente. Para este caso, el uso de un diagrama fuera de escala como el de la Figura 7 tiene justificación didáctica pues permite ubicar el punto triple metaestable dentro del diagrama general. No obstante, es indispensable señalar que no hay diferencia significativa entre las condiciones de aquel punto triple y del sistema estable a la temperatura Te. Luego, a pesar de la pequeña diferencia entre ellos permite que en determinadas situaciones y por razones estrictamente cinéticas, pueda alcanzarse el estado metaestable en lugar del más estable.
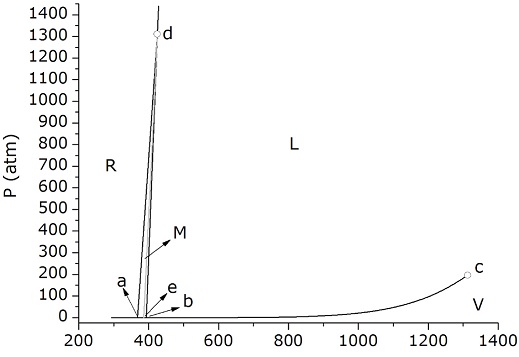
Figura 9a Diagrama de fases del azufre a escala real. La línea gris corresponde al equilibrio metaestable R-L.
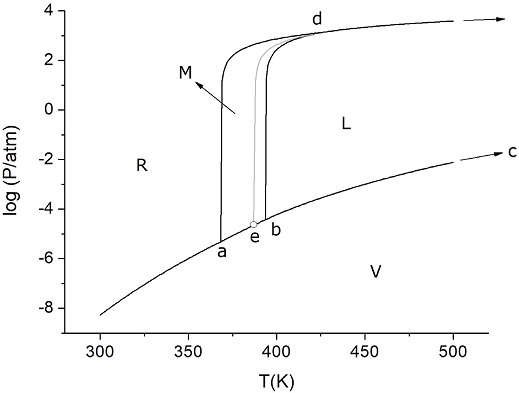
Figura 9b Diagrama de fases con escala logarítmica. La línea gris corresponde al equilibrio metaestable R-L.
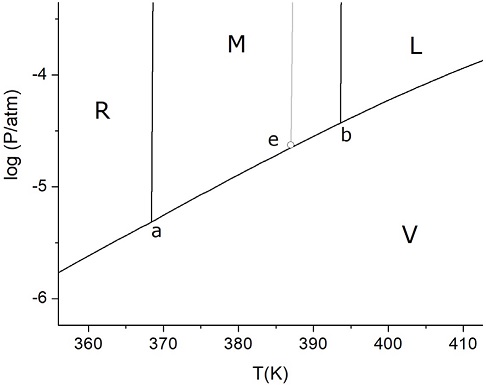
Figura 9c Detalle de la Figura 9b que muestra la proximidad del punto triple metaestable e, con la curva de equilibrio M-L.
Finalmente, se observa, tal como se anticipó cualitativamente en la Figura 3, que las curvas de equilibrio entre las fases condensadas y el vapor, que en el gráfico 9a tienen concavidad hacia arriba, manifiestan una concavidad hacia abajo en el diagrama semilogarítmico de la Figura 9b. Esta es una cuestión puramente matemática. La curvatura de una función está dada por su segunda derivada: si es positiva, la concavidad es hacia arriba; si es negativa su concavidad es hacia abajo. La expresión más simple para la presión de vapor de una fase condensada es (ver ecuaciones 2 y 3)
El signo de la derivada segunda respecto de T dos veces de la ecuación 11 depende de la diferencia B - 2T. Por lo tanto, la curvatura será positiva si T < B/2. De las ecuaciones 2 y 3, podemos verificar que esa temperatura es del orden de los 6000 K, por lo que la concavidad debe ser hacia arriba, tal como se observa en la Figura 4. Al aplicar logaritmos naturales a P, la expresión 11 se transforma en
Luego, la derivada segunda respecto de T dos veces es -2B/T3, por lo que será negativa para toda temperatura. De aquí que la concavidad de ln P sea hacia abajo.
Conclusiones
Aunque los diagramas de fase fuera de escala o con detalles exagerados para puntualizar ciertos comportamientos son, sin dudas, importantes desde el punto de vista didáctico, es posible que generen errores conceptuales (algunos de los cuales podrían ser serios. Esto se debe al fuerte impacto visual que tienen las representaciones gráficas, que graban aquellos detalles como si fueran un reflejo directo de un diagrama en escala real.
Algunos errores se inducen por las distorsiones que implican los diagramas esquemáticos, pero también, ciertos descuidos en la confección de diagramas a mano alzada pueden aún señalar comportamientos incorrectos (y que son gráficamente evitables).
En este trabajo hemos discutido algunos ejemplos de estos errores que se generan en torno a la descripción del diagrama de fases del azufre. El análisis presentado, puede tomarse como guía para una actividad de discusión en el aula, basada en la ecuación de Clapeyron y reservando, quizás para una asignatura más avanzada, el desarrollo del cálculo termodinámico.
Creemos que es relevante que los docentes adviertan y discutan estos inconvenientes con sus alumnos para hacer más adecuado el aprendizaje de un tema importante como el de los diagramas de fase, aunque también ciertos aspectos gráficos pueden generalizarse para cualquier otro contenido temático.

