











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkSobre la cristalografía de rayos-X y el nacimiento de la biología estructural
Desde los griegos, los científicos sospechaban que la naturaleza estaba compuesta por diminutas piezas a las que denominaron átomos. A comienzos del siglo XX se aceptó que los átomos existían y eran capaces de unirse entre sí. Las estructuras resultantes, que se denominaron moléculas, tenían propiedades químicas y físicas determinadas por su estructura atómica específica. Se habían desarrollado técnicas experimentales para averiguar la composición y la disposición de los átomos que formaban una molécula, pero nadie había visto el aspecto real de una molécula. Actualmente, cuando las moléculas cooperan y se estabilizan en forma de cristal, con un aparato llamado difractómetro de rayos-X y unos días de trabajo, es posible determinar la estructura tridimensional de la molécula.
Durante mis estudios de doctorado trabajé en la descripción estructural de compuestos inorgánicos determinados a partir de la técnica de difracción de rayos-X (Sánchez-Lara et al., 2018, 2019). Estas moléculas―que me permitieron obtener el grado de doctor en química―contienen poco más de 200 átomos y un peso molecular relativamente alto a causa de contener iones metálicos. Un ejemplo de estas moléculas “cristalográficamente determinadas” se muestra en la Figura 1 representada con Mercury, uno de los modernos y sencillos programas que permite la visualización, la exploración y el análisis de pequeños sistemas moleculares (Macrae et al., 2020). La cristalografía de rayos-X es una técnica fascinante porque nos permite ver la molécula, es decir, la manera en cómo los átomos se enlazan y están dispuestos en el espacio tridimensional. El único factor limitante de esta técnica es que la muestra a estudiar debe cumplir un requisito indispensable: ser cristalina.
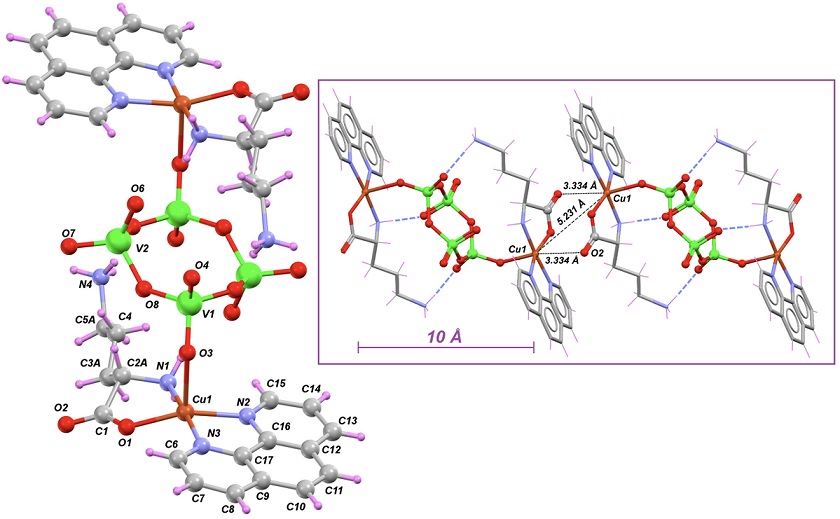
Figura 1 Estructura cristalina del compuesto {[Cu(Orn) (phen)]2[V4O12]}·6H2O obtenida a través de la cristalografía de rayos-X y representada con Mercury 4.0, una poderosa herramienta de visualización 3D (Macrae et al., 2020). En el recuadro se muestra el arreglo de las moléculas en el cristal (código de colores: V: verde; O: rojo; N: azul; C: gris; H: magenta). Para mayor información sobre detalles experimentales y descripción estructural ver Sánchez-Lara et al., 2021.
En un cristal―contrario a lo que ocurre en un líquido o un gas―las moléculas se encuentran dispuestas de manera ordenada, algo así como los árboles de un huerto plantado regularmente, y esta característica les concede la propiedad de difractar el haz de rayos-X. El azúcar, los increíbles copos de nieve o el chocolate―el cual consiste en uno de los cinco polimorfos del cacao cristalizado―son ejemplos de materiales cristalinos presentes en nuestra vida diaria. Entonces, cuando un haz de rayos-X incide sobre un cristal, los electrones que forman la parte externa de los átomos, dispersan o reflejan los rayos-X generando un patrón de difracción determinado, a través del cual―con la ayuda de una teoría matemática basada en la síntesis de Fourier―podemos resolver la estructura 3D de nuestro material cristalino. Con la estructura cristalina ya resuelta, podemos conocer de cerca a nuestra molécula. En el recuadro de la Figura 1 pueden verse algunos números. Indican la distancia que existe entre los átomos; podemos recopilar incluso ese detalle métrico.
Dentro del mundo biológico, las proteínas también pueden ser persuadidas a cristalizar para ser estudiadas a través de la cristalografía de rayos-X. Las proteínas son las moléculas activas de la vida. Cada molécula de proteína está conformada por una secuencia y combinación particular de un alfabeto de 21 aminoácidos que establecen su forma final. Las funciones específicas de las proteínas―que van desde catalizar las reacciones bioquímicas hasta la fabricación de los componentes estructurales básicos de los sistemas biológicos―van a depender esencialmente de su estructura 3D. Únicamente cuando adoptan esta configuración adquieren actividad biológica.
La rama de la biología molecular que se encarga del análisis estructural de las macromoléculas biológicas se conoce como biología estructural, y es un campo relativamente nuevo. El nacimiento de la biología estructural ocurrió en 1959 cuando John Kendrew, profesor en la Universidad de Cambridge, atacó con rayos-X cristales de mioglobina de ballena, una molécula de aproximadamente 2600 átomos (Chadarevian, 2018). La mioglobina es una proteína globular roja presente en el músculo esquelético de los mamíferos1 y al igual que la hemoglobina, se combina reversiblemente con el oxígeno (O2). No obstante, mientras que el papel de la hemoglobina es transportar el oxígeno de los pulmones a las células, el de la mioglobina es almacenarlo temporalmente en los tejidos. Algunas razones por las que Kendrew eligió trabajar con esta molécula fue por su tamaño relativamente pequeño y su tendencia a cristalizar fácilmente (Figura 2a).
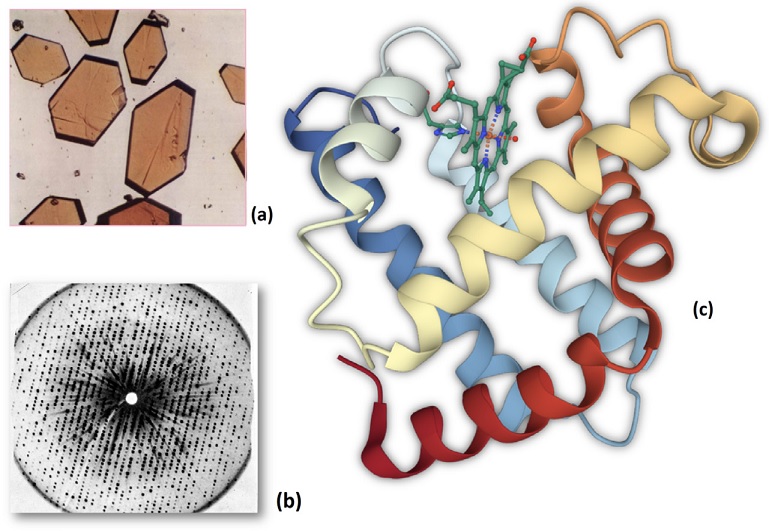
Figura 2 (a) Cristales de mioglobina preparados a partir de músculo de ballena; en ellos, las moléculas de proteína se encuentran ordenadas en un patrón regular. (b) Al dirigir un haz de rayos-X sobre un cristal y analizar el patrón de difracción obtenido, John Kendrew y sus colaboradores fueron capaces de graficar la densidad electrónica de la molécula y localizar la disposición de los átomos en el cristal (Chadarevian, 2018). (c) Estructura cristalina de la molécula de mioglobina (Entrada PDB: 1mbn), visualizada con Mol* (Sehnal et al., 2018); el grupo hemo se encuentra en la parte superior resaltado en color verde, en donde se observa cómo una histidina lo conecta con la proteína. Esta representación de la estructura proteica no detalla los átomos o los aminoácidos, pero refleja la disposición de la proteína.
Sin embargo, en la época del nacimiento de la biología estructural, no existía un programa de computadora (parecido a Mercury) que arrojara de forma rápida un retrato tridimensional y certero de la proteína a partir de los datos cristalográficos colectados. Por lo tanto, Kendrew construyó un enorme modelo de alambres de la mioglobina basado en los mapas de densidad electrónica obtenidos de los patrones de difracción (Figura 2b). Posteriormente, Irving Geis, un notable ilustrador científico, retrató la estructura de la mioglobina representando paso a paso, la enmarañada estructura 3D de la molécula. El resultado de este extraordinario trabajo científico y artístico se publicó en la revista de divulgación científica Scientific American bajo el título “The Three-dimensional Structure of a Protein Molecule”, en el cual, John Kendrew nos cuenta, con la emoción de quien ha descubierto un nuevo mundo, cómo determinó la arquitectura de esta proteína (Kendrew, 1961).
En la Figura 2c se muestra en toda su gloria tridimensional la estructura cristalina de la mioglobina, es decir, la manera en cómo los casi 2600 átomos que la conforman―que le confieren su identidad―están dispuestos en el espacio 3D. Es una imagen a la vez sorprendente y bella porque nos dice de manera precisa lo que somos a escala molecular.
Hablando en términos estructurales, la mioglobina es una “molécula pequeña” construida por una cadena polipeptídica de 153 aminoácidos, la mayoría de los cuales se encuentran organizados en ocho espirales o hélices-alfa; los aminoácidos restantes se emplean para unir estas cadenas helicoidales. En la superficie de la proteína podemos observar un objeto plano que parece no formar parte del paisaje proteico. Es un grupo hemo, una molécula en forma de anillo que contiene un átomo de hierro en el centro (ver Figura 2c). Este grupo no proteico o prostético, tiene la función de unirse de forma reversible con el oxígeno. El átomo de hierro del grupo hemo está enlazado a los cuatro átomos de nitrógeno de la molécula de porfirina, a una histidina llamada distal (his93) y a una molécula de oxígeno. Por lo tanto, el Fe se encuentra en un ambiente de geometría octaédrica. Conocer la geometría del grupo prostético es importante para poder entender la dinámica de la proteína al unir y liberar el oxígeno, una función que en la hemoglobina es particularmente importante.
Breve historia e impacto del Protein Data Bank
La determinación de la estructura atómica de la mioglobina a través de la cristalografía rayos-X sentó las bases de una nueva era de comprensión biológica. A partir de este hecho, se comenzaron a resolver casi simultáneamente las estructuras cristalinas de una serie de macromoléculas de enorme interés biológico; como la proteína hemoglobina, la lisozima y el ácido desoxirribonucleico (ADN). Para 1970, al menos una docena de estructuras biológicas habían sido resueltas a través de la técnica de difracción de rayos-X. Sin embargo, la tarea de compartir los archivos que contenían las coordenadas atómicas de los experimentos de difracción era lenta y complicada. En aquella época se empleaban cintas magnéticas para el almacenamiento de datos o tarjetas perforadas, en donde cada tarjeta representaba la información recogida de las coordenadas de un único átomo (Berman, 2020). Por lo tanto, intercambiar la información de la proteína mioglobina requería más de 2000 tarjetas, y hasta entonces, la única vía para difundir esta información era el correo postal.
Bajo estas circunstancias, en Junio de 1971 se celebró un histórico simposio organizado por el biólogo James Watson, en el Cold Spring Harbor, titulado “Structure and Function of Proteins at the Three-Dimensional Level” (Cold Spring Laboratory Press, 1972). En esta reunión se describieron de una forma tal las primeras estructuras de proteínas por los pioneros de la cristalografía macromolecular, que llevó a David Phillips a anunciar la mayoría de edad para la biología estructural. También hubo un intenso debate sobre la manera de intercambiar los datos cristalográficos. Entre los asistentes al simposio se encontraba Walter Hamilton, un tenaz cristalógrafo de moléculas biológicas pequeñas, quien se ofreció a albergar al Protein Data Bank en el Brookhaven National Laboratory (Berman, 2008). El establecimiento oficial del PDB fue anunciado en Octubre de 1971 como una nota en Nature New Biology resaltando que the succes of the proposed system will depend on the response of the protein crystallographers suplly data (Protein Data Bank, 1971). En este anuncio también se mencionaba la colaboración directa con el Cambridge Crystallographic Data Centre (CCDC) ubicado en Reino Unido a través de su fundadora Olga Kennard, quien tenía una enorme experiencia en archivar datos cristalográficos. El PDB se estableció con apenas siete estructuras cristalográficas.
En 1998, la gestión del PDB se volvió responsabilidad del Research Collaboration for Structural Bioinformatics (RCSB) y en 2003, en reconocimiento del crecimiento internacional y de la naturaleza interdisciplinaria de la biológica estructural, tres organizaciones unieron su energía para formar el Worldwide Protein Data Bank (wwPDB), con el propósito de mantener un único archivo de datos estructurales de macromoléculas biológicas (Berman et al., 2003). El RCSB, el Macromolecular Structure Database (MSD) y el Protein Data Bank Japan (PDBj) actúan como custodios del wwPDB asegurando que el PDB se encuentre disponible de manera gratuita y pública para la comunidad mundial.
A cincuenta años de su nacimiento, el crecimiento del PDB ha sido explosivo, en el momento de escribir este artículo hay poco más de 170,000 estructuras biológicas determinadas experimentalmente (de proteínas, ADN, ARN y sus complejos con fármacos u otras moléculas pequeñas) a disposición de los usuarios (Figura 3). De estas estructuras biológicas, alrededor de 154,000 han sido determinadas estructuralmente a través de la cristalografía de rayos-X. Las estructuras restantes, cerca del 12%, han sido determinadas a través de otras técnicas como Resonancia Magnética Nuclear (RMN) o Microscopia electrónica tridimensional (3DEM). En 2019, alrededor de 800 millones de coordenadas atómicas y archivos de datos experimentales fueron descargados con propósitos de investigación y educación (Worldwide Protein Data Bank, 2021). Estas descargas también incluyen las realizadas de forma rutinaria por empresas farmacéuticas y biotecnológicas para su uso en los esfuerzos de descubrimientos de nuevos fármacos.
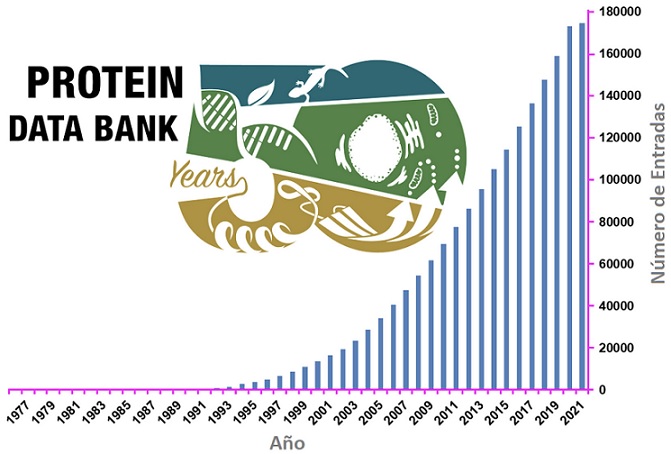
Figura 3 PDB en números: estructuras biológicas disponibles en el archivo hasta el 16 de Febrero del 2021. El número exacto para esta fecha es 174,994 macromoléculas biológicas depositadas (Protein Data Bank, 2021). También se muestra el logo conmemorativo que festeja los 50 años del Protein Data Bank.
Un estudio reciente en este aspecto ha mostrado que durante los años 2010-2016, el descubrimiento y desarrollo de 210 nuevas entidades moleculares aprobadas por la FDA (siglas para US Food and Drug Administration) han sido favorecidos tanto por la información estructural 3D generada por biólogos estructurales de todo el mundo, como por el carácter de acceso abierto del PDB, lo que ha permitido la identificación de blancos moleculares de manera eficiente (Westbrook y Burley, 2019; Goodsell et al., 2020). De estas nuevas entidades moleculares, 45 son moléculas de bajo peso molecular destinadas a terapia antineoplásica, cuyos blancos moleculares fueron identificados previamente con la ayuda del PDB.
Además de ser el único repositorio de datos biológicos estructurales, el RCSB PDB contiene un conjunto enorme de herramientas y recursos educativos sin restricciones. Entre estos servicios se encuentran los poderosos sistemas de visualización molecular que permiten superar las limitaciones del ojo humano. Uno de estos programas es el visualizador molecular Mol*, un proyecto reciente cuyo objetivo es ofrecer una biblioteca en común, y una serie de herramientas para la visualización 3D y el análisis de datos macromoleculares (Sehnal et al., 2018). Otro recurso importante es el portal en línea para profesores, estudiantes y público no especializado llamado PDB101 (http://pdb101.rcsb.org/), el cual promueve la exploración del fascinante mundo de las proteínas y de los ácidos nucleicos a través de una serie de materiales educativos, como modelos de papel (entre los que se incluyen el de la doble hélice del ADN, del virus Zika, de la hormona insulina, de la proteína verde fluorescente, de la cápside del VIH, entre otros.), animaciones interactivas, videos, libros para colorear, impresiones 3D, recursos para entender la pandemia del COVID-19 y algunos materiales adicionales para ampliar nuestro conocimiento sobre la biología estructural.
Una de las secciones más populares y de mayor tráfico del PDB-101 es la llamada Molécula del mes (Molecule of the Month), creada en el año 2000 por David S. Goodsell, en donde se destacan mensualmente las características estructurales y funcionales de una macromolécula biológica ubicada dentro del universo de datos del PDB y se discute su papel en el contexto de la salud y las enfermedades (Goodsell et al., 2015; 2020). Esta sección está destinada a tener un impacto en las actividades escolares y en el currículo. Por ejemplo, la molécula del mes destinada a anticuerpos hace referencia a la molécula de inmunoglobulina G (IgG) (Figura 4; Entrada PDB: 1igt), y emplea un fantástico modelo de papel para comprender la estructura y flexibilidad de esta biomolécula y por supuesto, su papel en el organismo. Este recurso puede emplearse para explicar la función que desempeñan los anticuerpos secuestrando patógenos como bacterias o virus, o en un curso introductorio sobre el sistema inmunitario.
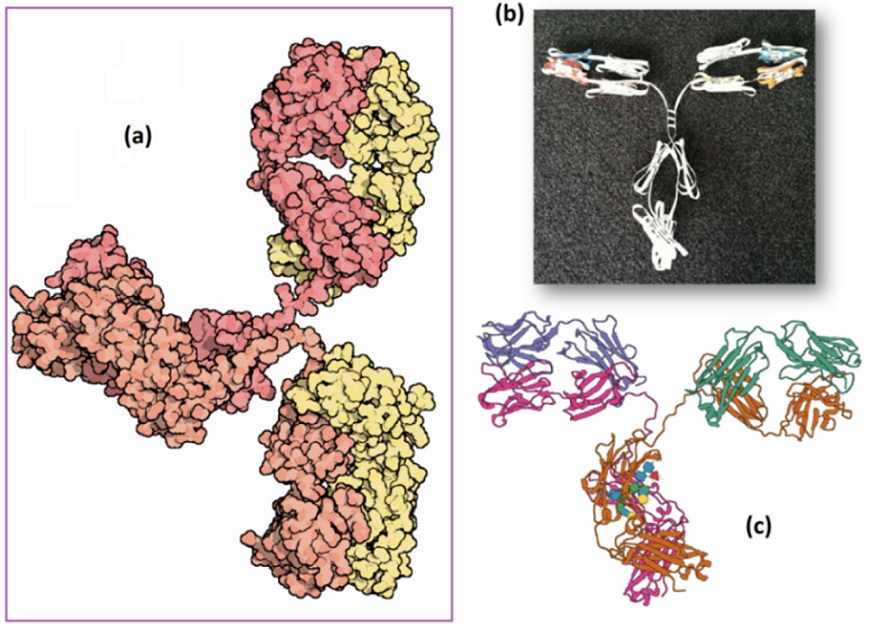
Figura 4 La forma sigue a la función: explorando la estructura de los anticuerpos. Imágenes obtenidas a partir de las herramientas que ofrece el portal PDB-101. (a) Vista de la inmunoglobulina G (IgG) con su típica estructura en forma de Y, en rojo se observan las cadenas pesadas con cuatro dominios Ig, mientras que en amarillo se muestran las cadenas ligeras. (b) Se puede utilizar un PDF descargable (junto con las instrucciones) para armar un bello modelo de papel del anticuerpo IgG. (c) La estructura tridimensional puede visualizarse empleando las coordenadas atómicas del PDB y puede compararse con el modelo de papel para correlacionar estructura química-actividad biológica.
Los virus ¿enemigos invisibles?
Los virus son un tema recurrente en el archivo del PDB debido a que constituyen una parte inseparable de la maquinaria básica de la vida y porque algunos tienen un efecto negativo en la salud humana.2 Un ejemplo es el virus de dengue o fiebre del dengue; una de las enfermedades víricas más importantes en términos de salud pública que se viven en estos tiempos. El virus se transmite fácilmente a los humanos por la picadura del mosquito tropical hembra Aedes aegypti. Anualmente se reportan de 300 a 500 millones de personas infectadas a nivel mundial, de las cuales 100 millones desarrollan la sintomatología (fiebre, nauseas, vómito y dolor general), 0.5 millones requieren hospitalización por complicaciones vinculadas a hemorragias graves, y alrededor de 20,000 mueren (Domachowske y Suryadevara, 2020).
A pesar de que la estructura 3D del virus del Dengue fue resuelta en 2002 a través de criomicroscopia electrónica y depositada en el archivo del PDB (Figura 5; Entrada PDB: 1k4r), todavía no existe un tratamiento específico para combatirlo a causa de que el virus presenta cuatro serotipos distintos (microorganismos infecciosos), cada uno con proteínas virales ligeramente distintas (Kuhn et al., 2002;). El dengue pertenece a la familia de los flavovirus, en la cual se encuentra el virus Zika, otro notable virus que se transmite por el mosquito A. Aegypti y que provocó una epidemia en el continente americano en 2016 relacionada a complicaciones neurológicas y malformaciones congénitas, conduciendo a la OMS a declarar a public health emergency of international concern (OMS, 2016). La estructura del Zika fue determinada en 2016 por criomicroscopia electrónica revelando semejanzas estructurales con el virus del dengue y otros flavovirus (Sirohi et al., 2016; Entrada PDB: 5ire).

Figura 5 Virus en estilo origami y mandalas del SARS-CoV-2. Ejercicios lúdicos de bajo costo realizados por estudiantes de cuarto y quinto de primaria empleando los recursos educativos que ofrece el PDB-101. Los estudiantes armaron el virus del dengue (a); del Zika con anticuerpos mostrados en azul (b), y de la cápside del VIH (c). También colorearon mandalas del coronavirus (d-f) identificando las principales proteínas virales: S, proteína spike; M, proteína de membrana; E, proteína de envoltura; N, proteína de la nucleocápside. Estas herramientas están disponibles de manera gratuita en el PDB-101 (http://pdb101.rcsb.org/), el portal educativo del RCSB PDB.
Conocer la estructura de los virus es esencial para entender su biología y emprender un camino a desarrollar vacunas para combatirlos. Por ejemplo, se ha estudiado el papel de un anticuerpo específico sobre el virus Zika que impide la entrada de material genético del virus a la célula. La estructura 3D de los anticuerpos anclados al virus ha sido reportada en 2016 y su estructura puede visualizarse en el archivo del PDB (Zhang et al., 2016; Entrada PDB: 5h37). Esta sorprendente estructura mostrada en la Figura 5b en estilo origami, muestra a las proteínas superficiales del virus y a los anticuerpos combatiéndolo.
Definir a los virus puede resultar controversial y ambiguo a la luz de los nuevos descubrimientos (Nasir et al., 2020). La National Human Genome Research Institute define virus como una partícula de código genético, ADN o ARN encapsulada en la vesícula de proteínas, los cuales no pueden replicarse por sí solos y necesitan secuestrar una célula con el fin de emplear su maquinaria molecular y hacer copias de sí mismos. A menudo, el virus daña o mata a la célula huésped en el proceso de multiplicación (NHGRI, 2021) Otros definen a los virus como organismos parásitos que viven en células infectadas y producen viriones para diseminar sus genes (Forterre y Prangishvili, 2009). Un virión es una partícula que puede ser purificada y visualizada. El núcleo de un virión comprende al ácido nucleico del virus (ADN o ARN) envuelto dentro de una cubierta proteica llama cápside (Nasir et al., 2020). El material genético del virus de la inmunodeficiencia humana (VIH) basado en ARN está envuelto en una enorme cápside en forma de cono con una simetría compleja, y lo libera en las células para infectarlas (Figura 5c). Debido al sorprendente tamaño de la estructura de la cápside del VIH su descubrimiento representó un avance significativo en la biología estructural, y un hito para el PDB cuando sus estructuras se depositaron en el archivo (Zhao et al., 2013; Entradas PDB: 1e6j; 3J3Q; 3J3Y).
Algunos virus conocidos como zoonóticos pueden dar un brinco fácilmente de los animales a los seres humanos y expandirse geográficamente a un ritmo alarmante, produciendo brotes epidémicos y pandemias. Este es el caso de los coronavirus causantes del síndrome respiratorio agudo severo (SARS-CoV) y del síndrome respiratorio de Oriente Medio (MERS-CoV). Dos virus patogénicos y altamente transmisibles que surgieron a comienzos del siglo XXI. Tanto el SARS-CoV como el MERS-CoV se trasmitieron directamente a humanos de mercados de civetas en China y de camellos dromedarios en el Oriente Medio, respectivamente (Cui et al., 2019; Schoeman et al., 2019). Estos estudios han mostrado que posiblemente el huésped natural del SARS-CoV es el murciélago de herradura (género Rhinolophus) y que la civeta simplemente actuó como un huésped intermediario. En el caso del MERS-CoV se tiene registro de que este virus tiene características genéticas vinculadas con otros coronavirus proveniente de murciélagos, y que su origen puede deberse a un intercambio de elementos genéticos entre ancestros virales, que incluyen a los aislados de camellos dromedarios y de murciélagos.
La maquinaria bioquímica que los coronavirus emplean para infectar a la célula se ha logrado identificar a partir de la cristalografía molecular, además de otras técnicas modernas (John et al., 2015; Schoeman et al., 2019). Los coronavirus contienen un genoma compuesto por una hebra de ARN de unas 30 mil pares de bases de longitud, una de las más largas de todos los virus de ARN. Este genoma se asemeja funcionalmente a un ARN mensajero cuando infecta una célula, orquestando la síntesis de poliproteínas que incluyen la maquinaria que el virus requiere para replicarse (Lubin et al., 2020); entre las cuales se se encuentran un complejo de replicación/transcripción que genera más ARN, varias proteínas estructurales que producen nuevos viriones, y algunas proteasas. Las proteasas juegan un papel esencial en fragmentar las poliproteínas en todas estas piezas víricas funcionales.
Al parecer, el surgimiento de los virus SARS en 2002-2003 en la provincia China de Guangdong y MERS diez años después en ciudades del Oriente Medio, no causó mayor preocupación en los gobiernos en turno ni en las organizaciones mundiales de salud para emprender medidas sanitarias de prevención y desarrollar recursos científicos para combatirlos. Y así, a finales del 2019, en un “mercado húmedo” de Wuhan, China, un nuevo coronavirus causante del síndrome respiratorio agudo severo (SARS-CoV-2) o COVID-19, migraba de un animal a un ser humano, permitiendo su transmisión y causando un colapso sin precedentes en los sistemas de salud y en la economía mundial (Wang et al., 2020). Hasta el día de hoy, 27 de febrero de 2021, a nivel mundial, la OMS ha reportado un total de 114 millones de casos confirmados y 2.52 millones de fallecidos. En México se han reportado 2 millones de casos confirmados y 185,000 mil fallecidos (Dong et al., 2020).
Un mirada a la estructura tridimensional de la pandemia
Al igual que los virus SARS y MERS, el SARS-CoV-2-agente causal de la pandemia de COVID-19-es un miembro de la familia de betacoronavirus de ARN que causa enfermedades en mamíferos y aves. Su material genético está escondido y protegido por proteínas que forman la nucleocápside debido a que es el actor protagónico de una serie de eventos moleculares básicos para que el virus prospere. Tan pronto infecta una célula, el virus se encarga de codificar una veintena de proteínas para replicarse (Lubin et al., 2020). La artillería que el SARS-CoV-2 utiliza para infectar una célula huésped se puede resumir de manera general en una polimerasa viral dependiente de ARN, proteínas Spike y enzimas proteasas. Estos sistemas moleculares que constituyen parte de la anatomía del virus, son blancos terapéuticos perfectos para combatirlo (Kearns, 2020). Por lo tanto, su identificación 3D por los biólogos estructurales ha sido una pieza clave para el desarrollo de una potencial vacuna.
Las proteasas son enzimas que fragmentan una proteína desenganchando un aminoácido de un extremo de la cadena polipeptídica. Se especializan―como lo hacen las proteínas― en ciertos aminoácidos. Un ejemplo de proteasas son la tripsina y la carboxipeptidasa A, enzimas digestivas que todos los animales, entre ellos los seres humanos, emplean para descomponer moléculas grandes para ser reformadas en moléculas mayores y mejores. En el caso de SARS-CoV-2 las proteasas juegan un papel esencial al fragmentar las poliproteínas en polipéptidos virales individuales, que son esenciales para la replicación y transcripción del virus, convirtiendo a las proteasas en un blanco atractivo biológico para el SARS-CoV-2. La estructura 3D de la principal proteasa (3CL o pro/Mpro) se publicó con un nuevo inhibidor en abril del 2020 y su estructura se encuentra depositada en el archivo del PDB con la entrada 6LU7 (ver Figura 6; Jin et al., 2020). La proteasa 3CL es un dímero de dos subunidades idénticas que juntas forman dos sitios activos. El plegamiento de la proteína es similar al de la proteasa serina (tripsina) pero los aminoácidos involucrados en el corte son la cisteína y la histidina. Un dominio extra estabiliza el dímero.
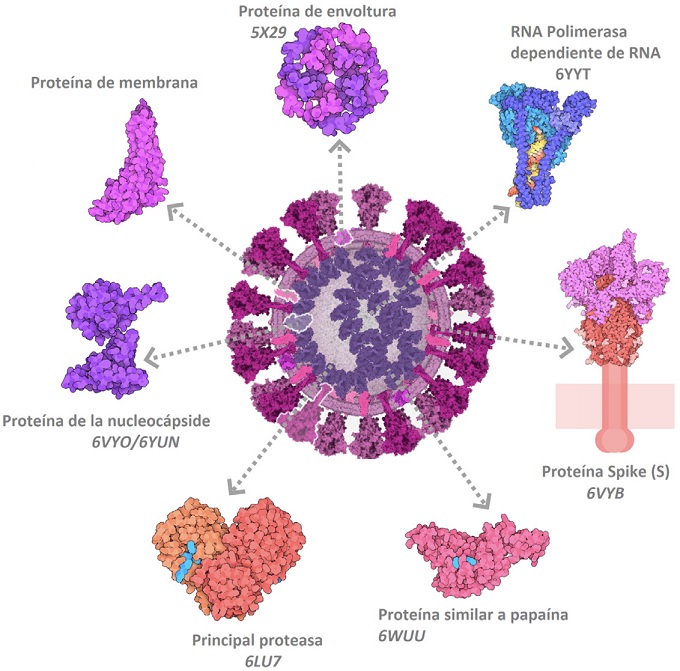
Figura 6 Estructura de la pandemia. Principales proteínas virales del SARS-CoV-2 con su código de entrada del PDB. Las imágenes fueron obtenidas de los recursos educativos del PDB-101, los cuales están basados en la evolución del proteoma del coronavirus durante los primeros seis meses de la pandemia por COVID-19 (Lubin et al., 2020).
La proteasa similar a la papaína (PLpro) es otra enzima esencial para la replicación y transcripción de los coronavirus, haciendo tres cortes específicos para procesar las largas poliproteínas del SARS-CoV-2. Una de las características de esta proteasa es su capacidad de sustraer la ubiquitina de algunas proteínas. La ubiquitina es una mini proteína responsable del reciclaje de moléculas biológicas obsoletas. Una de las consecuencias de esta eliminación es que interfiere en la producción de interferones, los cuales alertan a la célula de la presencia de agentes extranjeros (como los virus) para combatirlos (Baez-Santos et al., 2015). La disminución de la vigilancia viral permite al virus evadir la respuesta inmune de la célula huésped. Los inhibidores específicos de la PLpro como los basados en naftaleno afectan el efecto citopático que induce el virus, manteniendo la via de interferón antiviral, y reduce la replicación viral en las células infectadas (Shin et al., 2020). Actualmente, el PDB alberga 237 estructuras 3D de proteasas del SARS-CoV-2 esperando a ser exploradas.
Los coronavirus como el SARS-CoV-2 están basados en una larga hebra de ARN, la cual necesita replicarse dentro de la célula para el ensamble y liberación de nuevos virus. Por lo tanto, cuando el virus infecta una célula, codifica la síntesis de un complejo molecular que se encarga de catalizar la replicación de ARN. Este complejo está formado por proteínas no estructurales (nsps) y por un componente central que recibe el nombre de ARN Polimerasa dependiente de ARN. Este sistema emplea ARN como plantilla―y no ADN―para su replicación. La arquitectura de este extraordinario complejo ha sido completamente determinada (Figura 6; Entrada PDB: 6YYT) tanto en la forma apo (inactiva) como en su complejo con ARN y con el fármaco antiviral remdesivir, uno de los fármacos aprobados para su uso de emergencia contra el SARS-CoV-2 (Yin et al., 2020). El hecho de que la replicación está basada en un plantilla de ARN, hace que este complejo sea un blanco específico para combatir el virus debido a que difiere estructuralmente de las proteínas de la célula hospedera.
Las proteínas que se encuentran en la parte externa del SARS-CoV-2 y que le dan el aspecto de corona solar, son las glicoproteínas Spike o proteínas S, responsables de comunicarse con los receptores de la célula huésped y favorecer su internalización dentro de ella. Al igual que las proteasas y la ARN polimerasa, la proteína Spike es un objetivo importante para vacunas y anticuerpos terapéuticos que logren bloquear su comunicación con los receptores celulares. Se ha descubierto que una de las dos subunidades (S1) que conforman esta proteína estructural contiene un dominio de unión al receptor (llamado RBD por Receptor Binding Domain), el cual tiene una alta afinidad por la proteína transmembranal convertidora de angiotensina humana (ECA2).
La ECA2 es una enzima que activa a la angiotensina, una hormona peptídica que participa en la vasoconstricción y en el control de la presión sanguínea. ECA2 es expresada en células de pulmón, corazón, riñón y de intestino, haciéndolas blancos idóneos para ser colonizadas por el virus. La otra subunidad (S2) se encarga de fusionarse con la membrana de la célula huésped para lograr la entrada del virus. Sorprendentemente, en el 2020 se logró determinar por cristalografía de rayos-X la estructura 3D de la glicoproteína Spike (Wrapp et al., 2020; Entrada PDB: 6VXX) y del receptor celular ECA2 (Lan et al., 2020; Entrada PDB: 6M18). Además se logró determinar la manera en que ambas macromoléculas (viral y humana) se reconocen entre sí, revelando las bases estructurales que emplea el SARS-CoV-2 para iniciar el proceso de infección (Yan et al., 2020; Entrada PDB: 6M17).
Se ha revelado (teóricamente) que algunos compuestos de vanadio tienen capacidad de inhibir a la principal proteasa del SARS-CoV-2, la proteasa 3CL (Scior et al., 2021). Me pregunto si el compuesto que se muestra en la Figura 1, tendrá alguna actividad inhibitoria sobre las moléculas biológicas del virus discutidas aquí.
Observaciones finales
En 1962, John C. Kendrew compartió con Max Perutz el premio Nobel de Química por sus estudios de las estructuras de proteínas globulares. El premio Nobel de Fisiología estuvo destinado el mismo año a James Watson y Francis Crick por haber determinado, a través de la cristalografía de rayos-X, la estructura tridimensional del ADN. Estos personajes, junto con las moléculas que determinaron fueron los protagonistas de la película molecular de la vida. Con el tiempo, la acumulación de un conjunto de datos estructurales de macromoléculas biológicas dio origen al Protein Data Bank. El PDB cumple 50 años en un momento crítico, en donde se hace evidente que los elementos más importantes de nuestro mundo globalizado dependen en gran medida de la ciencia y la tecnología. En este sentido, la biología estructural ha jugado un papel esencial en responder al reto más importante de lo que va del siglo XXI: el SARS-CoV-2.
Parece que Sun Tzu mencionó en el arte de la guerra que los enemigos que se conocen son más fáciles de combatir. Las vacunas que se han desarrollado para luchar contra la pandemia son el resultado del conocimiento científico acumulado en duros meses de trabajo. Estudios que han permitido comprender los mecanismos biológicos por los que el SARS-CoV-2 infecta una célula. La ciencia tiene rostro, y estamos en deuda con los biólogos y biólogas estructurales que han trabajado a marchas forzadas para conocer los planes de un enemigo de 100 nanómetros (Bárcena et al., 2021). El PDB no ha sido ajeno a este reto y se ha sumado a la lucha contra la pandemia. En su archivo se han depositado casi 1000 estructuras del nuevo coronavirus esperando a ser exploradas. Además, el portal cuenta con recursos educativos para ayudar a comprender la pandemia desde un punto de vista científico. Actualmente, el RCSB PDB contiene más de 170,000 entradas y en el año pasado la comunidad mundial descargó más de 800 millones de datos. Lo que era un sueño en 1971, se ha convertido en una herramienta esencial de la biología moderna

