










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de neurociencias (México, D.F.)
versión On-line ISSN 1028-5938versión impresa ISSN 0187-4705
Arch. Neurocien. (Mex., D.F.) vol.10 no.2 Ciudad de México abr. 2005
Artículo original
Alteraciones neuro–oftalmológicas en pacientes que presentan enfermedades mitocondriales
Neuro ophtalmological alterations in mitocondrial diseases
Irene González–Olhovich, David Lozano Elizondo
Instituto Nacional de Neurología y Neurocirugía Manuel Velasco Suárez Departamento de Neuro–oftalmología.
Correspondencia:
David Lozano–Elizondo.
Departamento de Neuro–oftalmología Instituto Nacional de Neurología y Neurocirugía.
Insurgentes Sur # 3877. Col. La Fama
14269 México, D.F.
Recibido: 2 febrero 2005.
Aceptado: 18 febrero 2005.
RESUMEN
Las enfermedades mitocondriales según se ha demostrado tienen variadas alteraciones neuro–oftalmológicas siendo las más frecuentes atrofia óptica oftalmoplejía, retinopatía pigmentaria y alteraciones retroquiasmaticas. Se estudiaron 23 pacientes con diagnóstico de enfermedad mitocondrial por biopsia muscular y con diversas alteraciones oftalmológicas un 69.6% con disminución de agudeza visual. Por el momento no hay tratamiento efectivo si es muy importante el diagnóstico pronóstico y no usar tratamientos inútiles.
Palabras clave: enfermedad mitocondrial, alteraciones oftalmológicas, pronóstico, tratamiento.
ABSTRACT
Mitocondrial diseases produce a series of neuro–ophthalmological diseases. The must comon, optic atrophy, oftalmoplegia, pigmentary retinopaty and retroquiamatic alterations. Twenty three patients with mitocondrial disease diagnosed by muscle biopsy were studied, 69.6% had decrease of visual acuity. No tratment is available at the moment but the diagnosis is basic for prognosis and avoid unnecessary treatments.
Key words: mitocondial diseases, alterations treatment ophtalmological, alterations.
Se han observado en numerosos estudios la asociación de enfermedades mitocondriales con manifestaciones neuro–oftalmológicas. Las citopatlas mitocondriales han tenido en los últimos años una gran importancia, anteriormente la neuropatía hereditaria de Leber se ha reportado, con tres genes asociados a herencia autosómica dominante, al igual que la Oftalmoplejía progresiva externa.
Las anormalidades más frecuentes en las patologías mitocondriales son la atrofia óptica bilateral, oftalmoplejía con ptosis, retinopatía pigmentaria y alteraciones retroquiasmáticas.
Debido a que las enfermedades mitocondriales son un grupo de patologías de gran interés y presentan una gran variedad de diferentes tipos de presentaciones sistémicas, es de suma importancia tener las manifestaciones oftalmológicas presentes, en el caso de observar una neuropatía óptica sin explicación, una oftalmoplejía sin otros datos que provoquen alteraciones retinianas o campimetría retroquiasmatica sin otra explicación, se tiene que tener en mente la posibilidad de que el paciente presente una citopatía mitocondrial y se debe de realizar una exploración minuciosa del paciente para dar un diagnóstico mas preciso.
Se propone con este estudio realizar una evaluación neuro–oftalmológica completa de los pacientes con el diagnóstico de citopatías mitocondriales en el Instituto Nacional de Neurología y Neurocirugía, para poder obtener un patrón de las características neuro–oftalmológicas que se presenten en nuestros pacientes.
ANTECEDENTES
Las mitocondrias son organélos celulares que influyen en múltiples reacciones bioquímicas, que tienen como finalidad la producción de energía. Las enfermedades mitocondriales son anormalidades hetereogénicas en términos bioquímicos, hereditarios, con fenotipos clínicos. Los síndromes neurológicos son los que presentan mayor afección ya que el cerebro y los músculos son los que presentan mayores demandas de energía1.
La primera correlación entre enfermedades mitocondriales y síndromes neurológicos se reportó por Olson 1972, en un paciente que presento fibras rojas rasgadas en músculo, con diagnóstico de Kearn Sayre, En 1988 Zeviani encontró las deleciones del ADN, en el mismo año Wallace describió una mutación del ADN que causa la Neuropatía Óptica de Leber. Un año después Perker, describe un defecto en el electrón de transporte mitocondrial en este síndrome. En 1951 se describe la enfermedad de Lieg, en 1980 se describen los de síndromes epilepsia con fibras rojas rasgadas (MERRF) y encefalopatía mitocondrial, acidosis láctica, episodios de Stroke–like (MELAS), correlacionándolos con herencia materna que posteriormente se asocian a mutaciones del ADN mitocondrial1–4.
Las enfermedades mitocondriales es un grupo de patologías multisistémicas heterogéneas en las cuales la presentación clínica; genética, bioquímica, e histopatológica presentan una disfunción mitocondrial. Las disfunciones mitocondriales resultan de anormalidades del ADN mitocondrial (mtADN) o de anormalidades estructurales de las proteínas nucleares de la mitocondria, presentando contribuciones de dos genomas, los cromosomas nucleares y los mitocondriales. Los diferentes grados de expresión de las anormalidades mitocondriales varian de enfermedad a enfermedad y de paciente en paciente. Presentan una herencia materna y el punto de mutación del mtADN se hereda por esta misma vía.
Las citopatías mitocondriales son difíciles de clasificar y se han realizado dos grupos basados en la presentación clínica y bioquímica, también se ha propuesto en base al análisis genético en el punto de mutación, deleción, depleción y duplicación, desafortunadamente hay una gran inconsistencia de la correlación entre la presentación clínica y los defectos genéticos.
Las manifestaciones neuro–oftalmológicas en las enfermedades mitocondriales se han observado cuatro presentaciones clínicas principales: la neuropatía óptica bilateral, la oftalmoplejía con ptosis, la retinopatía pigmentaria y las alteraciones campimétricas retroquiasmáticas.
En cuanto a la neuropatía óptica bilateral la patología más común es la atrofia óptica de Leber (LHON), esta patología fue de las primeras que se asoció a defectos en mtADN, presentando solo datos neuro–oftalmológicos. Su expresión es predominante en hombres, la susceptibilidad de la pérdida visual no se ha explicado, la edad de presentación es entre los 15 y 35 anos, pero el rango puede ser de 1 a 80 años. La pérdida visual es indolora, central y por lo general ocurre en un ojo y semanas o meses después en el segundo, la agudeza visual es 20/200 con defectos centrales o cecocentrales en los campos visuales (CV). La función pupilar se preserva, esto nos indica un daño diferente a la fibras aferentes pupilares en el nervio óptico, esto continúa en debate. En las fases agudas de la perdida visual se puede encontrar con una papila hiperemica, dilatación de los vasos, hemorragias, microangiopatía peripapilaro ligero edema papilar. En la mayoría de los pacientes con LHNO, la perdida visual es la única manifestación, aunque se ha observado en miembros familiares alteraciones en la conducción cardiaca, principalmente síndromes de prexcitación, algunas anormalidades neurológicas y en músculos. Los potenciales visuales evocados presentan anormalidad importante, la fluorangiografía ayuda para distinguir la alteración en nervio óptico de un edema verdadero, el electrorretinograma por lo general es normal, electroencefalograma, líquido cefalorraquídeo y pruebas de imagen no presentan datos de importancia, en la resonancia magnética en T2 se puede observar hiperintensidad en los nervios ópticos en algunos pacientes.
Se ha observado tres puntos de mutación en mtADN en los pacientes con LHON, la primera mutación que es la responsable del 90 al 95% se localiza en nucleótido mtADN posición 11 778 (69% de los casos), 3 460 (13% de los casos) y 14 484 (14% de los casos), otros puntos de mutación se han encontrado en menos frecuencia, 72% de los pacientes con mutación en 14 484 presentan menor alteración en la visión que el 5% de los pacientes con mutación en 11 778, los pacientes con mutación en 3 460 presentan mejor recuperación visual que la posición 11 778, pacientes mas jóvenes menores de 15 años presentan mejor pronóstico visual.
El diagnóstico de LHON se debe de considerar cuando no se observe ninguna causa para la atrofia óptica bilateral, la edad de presentación, historia familiar y la presentación clínica. Los defectos genéticos no se pueden del todo explicar en la expresión de la enfermedad. La presencia de mutación mtADN es necesaria para la expresión fenotípica pero no es suficiente. La presencia de heteroplasmia (co–existencia de mutación y mtADN normal), es un factor en la expresión.
La fisiopatología de LHON continúa sin saberse pero es probable que anormalidades en la fosforilación oxidativa y la deficiencia en la generación de A TP ya sea directa o indirectamente libera radicales libres, resultando un daño de las células ganglionares y sus axones, la razón por la cual los mecanismos selectivos solo al nervio óptico (NO) no se saben, pero se sugiere que en el ángulo que hacen las fibras los axones de las fibras nerviosas en la región prelaminar se crea un punto critico para el transporte axoplásmico.
Otras patologías mitocondriales con presentación clínica y genética se ha observado atrofia óptica como variante o manifestación secundaria, esto hace que sea la manera de distinguirlas de LHON en la cual es la primera y muchas veces única manifestación clínica, como ejemplo de estas otras patologías se encuentran casos en la epilepsia y fibras rojas rasgadas, encefalopatía mitocondrial, acidosis láctica y eventos vasculares isquémicos–like (MELAS), oftalmoplejía progresiva extema, síndrome de Leigh. La neuropatía óptica se debe de considerar como marcador para las enfermedades mitocondriales.
La oftalmoplejía progresiva externa (CPEO) es un grupo clínico que se caracteriza por progresión lenta bilateral de inmovilidad ocular asociada a ptosis. La oftalmoplejía y ptosis se asocian a otros datos neurológicos y sistémicos como miopatías generalizadas, sordera, ataxia, espasticidad, neuropatía periférica, miopatía, neuropatía gastrointestinal, disfunción vestibular, demencia, encefalopatía episódica y calcificaciones de ganglios básales.
Las presentaciones oculares se asocian atrofia óptica, retinopatía pigmentaria, cambios corneales, cataratas. El involucro sistémico se observan alteraciones cardiacas, endocrinas, alteraciones en pie, se presentan anormalidades en la conducción cardiaca, estatura corta, diabetes, retraso en la maduración sexual, hipogonadismo, hipomagnesemia, hipopara–tiroidismo e insuficiencia respiratoria.
El síndrome de Kearns–Sayre (KSS) es un subtipo de CPEO con alteraciones sistémicas y neurológicas el diagnóstico se basa en: 1. edad de presentación antes de los 20 años, 2. CPEO, 3. degeneración pigmentaria, 4. con uno o varios de los siguientes datos: anormalidades en la conducción cardiaca, elevación en las proteínas del líquido cefalorraquídeo (LCR) (más de 100 mg/dl), disfunción del cerebelo. La histopatología de cerebro presenta cambios espongiformes y en imagen se observan anormalidades que corresponden a calcificaciones de ganglios básales. La presencia de fibras rojas rasgadas se demuestran con la tinción tricrómica modificada, de los músculos en las extremidades y músculos extraoculares.
En el análisis de ADN de músculo esquelético de pacientes con CPOE se presentan desarreglos de segmentos de mtADN del tipo de deleción y duplicación, los desarreglos son heteroplasmicos. Los pacientes con KSS presentan un porcentaje mayor de mutación en los tejidos que los presentan CPEO. El desarreglo se observa en el 90% de los pacientes con KSS comparado con 50% en los CPEO. La mayoría de los casos de CPEO son esporádicos, los familiares pueden presentar el desarreglo pero no los datos clínicos ni alteraciones en la biopsia, se pudiera pensar que el desarreglo sucede durante la embriogenesis, aunque se han observado casos de herencia dominante con anormalidades nucleares del ADN, las mutaciones en tres genes diferentes ANTI (cromosoma 4q34–35), TWINKLE (cromosoma 10q24), POLG (cromosoma 15q22–26).
La CPEO es raro que se observe en pacientes con MELAS, pero puede estar en la encefalopatía neurogastrointestinal mitocondrial que se presenta como autosomica recesiva y clínicamente presenta dismotilidad gástrica severa, caquexia, ptosis, oftalmoparesia, neuropatía periférica y leucoencefalopatía, los pacientes se mueren en edades tempranas, la mutación es en el gen nuclear de fosforilasa tiamina en el cromosoma 13.32–q.
La retinopatía pigmentaria se observa en pacientes con mitocondriopatía, se presenta como retinopatfa en "sal y pimienta", es más prominente con la edad, puede presentarse involucro macular, atenuación vascular. Histopatológicamente se observa degeneración del epitelio pigmentario de retina, anormalidades en cono y bastones. La pérdida de visión ocurre en el 50% de los casos y es moderada. La fluorangiografía y el electroretinograma pueden confirmar los cambios.
La retinopatía pigmentaria es el mayor criterio diagnóstico del síndrome de KSS, se pueden observar degeneraciones pigmentarias de la retina en pacientes con CPEO sin otras alteraciones neurológicas.
La distrofia muscular se reporta en pacientes con mutaciones 3243, que es típico de MELAS en el síndrome de diabetes Mellitus y sordera. La retinopatía pigmentaria se presenta en pacientes con punto de mutación del mtADN en la posición 8993 en el gen de ATPase–6, esta mutación se describe como de linaje materno con retinopatía pigmentaria, retraso en el desarrollo, demencia, crisis convulsivas, ataxia, debilidad muscular proximal neurogénica y neuropatía sensorial. Esta misma mutación del mtADN se asocia también en un tercio de los pacientes con síndrome de Leigh, una encefalopatía de la infancia o de la juventud con retraso psicomotor, hipotonía, anormalidades de cerebelo, acidosis láctica, cambios espongiformes en encéfalo y ganglios básales, hipoventilación central respiratoria, nistagmus y alteraciones en movimientos oculares supranucleares así como atrofia óptica.
No se observa una correlación en la presencia de cambios retinianos con defecto genético de, anormalidad bioquímica y otros datos clínicos, pero la presencia de retinopatía pigmentaria en un paciente con enfermedad neurológica o sistémica debe de hacer la sospechar de citopatía mitocondrial.
Las alteraciones campimétricas retroquiasmáticas se observan en pacientes con mitocondriopatías que no son secundarias a alteraciones en el nervio óptico, disfunciones retinianas. Por lo general, se presentan como hemianopsias homónimas o ceguera cortical. La patología en la que mas se observan estas alteraciones es en MELAS, que es una patología que se caracteriza por ataques recurrentes abruptos de cefalea, vómito, convulsiones focales y generalizadas, síntomas neurológicos focales que duran horas o dias, hay una predilección por encéfalo posterior, las alteraciones visuales se reportan en la mitad de los pacientes. Las fibras rojas rasgadas se observan en el 90% de los pacientes, mitocondrias anormales se han observado en músculo liso de vasos sanguíneos, esto es de suma importancia para los eventos vasculares "stroke–like" en los pacientes con MELAS. En los estudios de imagen se observan calcificaciones de los ganglios básales y focales en la corteza cerebral, las lesiones se observan en sustancia blanca que representa un edema vasogénico, las lesiones desaparecen con el tiempo. Se presenta esporádicamente o puede ser familiar, con herencia materna, aproximadamente el 90% presentan una mutación del mtADN en la posición 3243 en el gen de transferencia.
La importancia del conocimiento de las mitocondriopatías sobre las alteraciones sistémicas y neurológicas es hoy en día de gran interés. Es necesario tener en cuenta y saber detalladamente cuales son los datos neuro–oftalmológicos en estas entidades, ya que las alteraciones oculares forman parte indispensable para el diagnóstico ya sea por la presencia o ausencia de los diferentes datos.
MATERIAL Y MÉTODOS
Se realizará un estudio prospectivo, longitudinal, observacional en, pacientes del Instituto Nacional de Neurología y Neurocirugía con el diagnóstico de enfermedad mitocondrial.
Se les realizó estudio neuro–oftalmológico completo incluyendo agudeza visual, pruebas cromáticas, examinación con lámpara de hendidura, exploración bajo dilatación pupilar con fármacos midriáticos para la revisión minuciosa de retina periférica, realización de toma de presión intraocular, campimetría de Goldmann, elecarretinograma, potenciales visuales evocados, resonancia magnética y/o tomografía computarizada.
Criterios de inclusión: pacientes que tengan expediente en el Instituto y que hayan sido valorados por el servicio de nervio y músculo del Instituto.
Pacientes con el diagnóstico de citopatología mitocondrial establecido: a. clínicamente b. biopsia muscular de cualquier género y edad.
Las variedades el estudio fueron, agudeza visual, pruebas cromáticas, campimetría de Goldmann, presión intraocular, medición de alteraciones en pacientes con ptosis, electroretinograma, potenciales evocados visuales, resultado de estudios de imagen.
Los instrumentos de medición:
Agudeza visual se tomó con tablas de Snellen, presión intraocular con termómetro de aplanación de Schotz, campos visuales con campímetro de Goldmann, exploración de retina, se estudió con lente de tres espejos, electrorretinograma, potenciales visuales evocados.
Para las variables demográficas se utilizaron medidas descriptivas de tenencia central y dispresión, así como proporciones para determinar la frecuencia de las diferentes alteraciones.
RESULTADOS
En este protocolo de investigación se realizó un estudio descriptivo de las alteraciones neuro–oftalmo–lógicas encontradas en pacientes que presentaron enfermedades mitocondriales con diferentes diagnósticos.
Se realizó estudio oftalmológico completo a un total de 23 pacientes con diagnóstico de citopatía mitocondrial basado en los hallazgos de la biopsia muscular (por la presencia de agregados mitocondriales, fibras "rojas rasgadas", acumulo de lípidos o material PAS positivo y/o fibras COX negativas) y su correlación clínica.
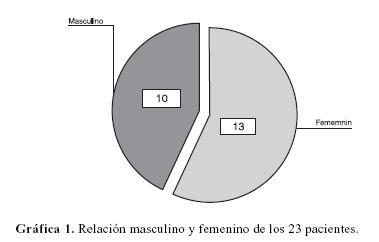
La muestra fue de un total de 13 pacientes femeninos (57%) y 10 masculinos (43%) (gráfica 1), con una media de edad de inicio de 8 años y un rango de 2 meses a 46 años, la edad de los pacientes al momento de la exploración neuro–oftalmológica fue en promedio de 30 años, (rango 17 a 55 años).
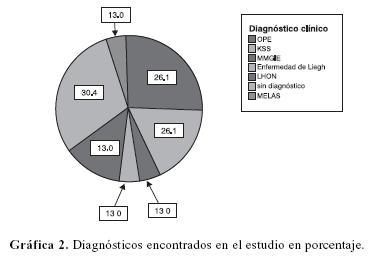
Los diagnósticos encontrados en el estudio fueron oftalmoplejía progresiva externa (OPE) en un 26.1%, síndrome de Kearn–Sayre (KSS) en un 17.4%, síndrome de encefalomiopatia mitocondrial neurogastrointestinal (MMGIE) 4.3%, enfermedad de Leigh (MILS) 4.3%, neuropatía óptica hereditaria de Leber (LHNO) en 13.0%; encefalomiopatia mitocondrial con acidosis láctica y episodios stroke–like (MELAS) en 4.3% no encontrando diagnóstico definitivo en 7 pacientes (30.4%) (gráfica 2).
Se dividieron en dos grupos en los cuales se clasificaron según la presencia o ausencia de manifestaciones extraoculares, encontrándose que solo 10 pacientes (43%) presentaron alteraciones oftalmológicas sin tener afección a otros órganos, en 13 pacientes se presentó la combinación de afecciones oftalmológicas y afección sistémica.
En cuanto los antecedentes heredo–familiares, encontramos que en el 47.8% de los pacientes si había historia familiar del padecimiento, mientras que en 52.2% no presentaban familiares afectados.
La disminución de agudeza visual (AV) se refirió como antecedente antes de presentar el cuadro completo de la enfermedad en 30.4%, 69.6% no refirieron.
En cuanto a la AV al momento de la exploración oftalmológica se tomó como mayor importancia la AV corregida en donde encontramos una AV ojo derecho (OD) de 20/20 en 43.5%, en ojo izquierdo (Ol) 47.8%; menor de 20/50, en ambos ojos en 7 pacientes, considerando como ceguera legal (< 20/100) 17.3% en ambos ojos, con alteraciones en las pruebas cromáticas en ambos ojos de 17.4%, encontrados en la normalidad en 82.6%.
Las presiones intraoculares en todos los pacientes se encontraron en rangos de normalidad. En cuanto los movimientos oculares encontramos que ptosis palpebral fue la primera manifestación de enfermedad 56.5%. En el momento de la exploración se observó una apertura palpebral menor de 10 mm en ambos ojos en el 56.5% de los casos, una función del elevador menor de 10 mm en OD en 69.4%, Ol 65%, encontrándose una nula función del elevador en 30.4% de ambos ojos, por consecuencia 56.5% presentaron un fenómeno de Bell negativo.
Sólo 8.7% de los pacientes refirieron diplopía desde el inicio del padecimiento, la cual se confirmó con exploración de lente rojo en solo un caso (4.3%).
Identificamos alteración en los movimientos oculares en 14 pacientes (60.8%), sorpresivamente el 95.7% refirieron no haberse dado cuenta de que presentaron alteraciones en los movimientos oculares. En cuanto a los pacientes que presentaron afección muscular al momento de la exploración oftalmológica; OD afección de más de 5 músculos 56.5%, Ol 52.2%.
El reflejo optocinético se encontró negativo en el 78.3% de los pacientes. Se encontró en ortoposición 34.8% de los pacientes y con un exodesviación simple en el 56.6%, compuesta en un 8.6%.
En exploración con la lámpara de hendidura se encontraron los siguientes resultados: conjuntiva normal en OD 82.6%, Ol 87.0%; córnea sin alteraciones en 82.6% en ambos ojos, iris regular en OD en el 95.7%, en Ol 91.3%; cristalino transparente OD 52.2%, Ol 56.5%, opacidades subcapsulares posteriores en 4.3% en ambos ojos, opacidades puntiformes en ambos ojos se observaron en 39.1%, un ojo con implante de lente intraocular (4.3%).
En la exploración bajo dilatación pupilar se encontraron los siguientes datos: papila con coloración anaranjada (considerada como normal) 52.2% en ambos ojos, palidez (+ a + + + +) 26% ambos ojos, atrofia papilar en 17.4% y un paciente con papilas hipoplásicas (4.3%); no se observaron alteraciones en los bordes neuroretinianos en ningún paciente; los vasos retinianos se encontraron centrales en 87% en ambos ojos, no encontrándose cambios en retina en 17.4% en ambos ojos, cambios centrales en el epitelio pigmentado de la retina OD 26. %, 0121.7%, cambios del epitelio pigmentado de la retina periférica en 39.1 % OD, 01 43.5%, espículas óseas se encontraron en un paciente (4.3%), fondo coroideo bilateral en 13%.
Los estudios realizados para ver función retiniana y de nervio óptico fueron los siguientes: electrorretinograma (ERG) anormal en el 47.8% y potenciales visuales evocados (PVE) anormales en el 52.2%. Las alteraciones del ERG fueron alteraciones en la integridad de los componentes a y b de latencia absoluta, amplitud, morfología y replicabilidad de los fotorreceptores, células ganglionares y cél de Müller en la retina; en cuanto las anormalidades que se encontraron los PVE fueron integración de un potencial cortical P100 de latencia prolongada, baja amplitud, moderada a severa cono dispersión y replicabilidad en las fibras del nervio óptico, localizándolas con mayor precisión fibras maculares o de retina periférica.
Los campos visuales se encontraron normales en 56.5%, disminución concéntrica en 17.4% y en el resto se encontró hemianopsias, escotomas cecocentrales, isla de visión, en dos pacientes no fueron valorables (8.7%).
DISCUSIÓN
El estudio de las enfermedades mitocondriales ha aumentado importantemente ya que la prevalencia de estas enfermedades en la población general no se conoce y esto promueve el interés por seguir investigando.
La población de este estudio fue de 23 pacientes, se encontró que 13 pacientes fueron femeninas y 10 masculinos, no siendo una diferencia significativa.
Los pacientes en este estudio presentaron una edad de inicio de los síntomas en un rango de meses a 46 años siendo la más frecuente 8 años (21.7%), por lo que nos indica que la edad de presentación es muy similar a los demás estudios en los que se cita que la edad de inicio es en la adolescencia (<20 años), teniendo en cuenta que en los diferentes síndromes pudiera variar en la edad de presentación.
Al momento de la valoración neuro–oftalmológica la mayoría de los pacientes ya contaban con un diagnóstico establecido, o ya habían acudido con diferentes facultativos por su tratamiento no teniendo un diagnóstico correcto en varios casos, lo que retardo su atención en nuestro servicio, teniendo un rango de 17 a 55 años.
Se encontró que en la población de nuestro estudio el nivel socioeconómico fue medio–bajo, lo cual pudiera estar relacionado al tipo de población que se estudia en el Instituto Nacional de Neurología y Neurocirugía. La escolaridad de nuestra población fue en su mayoría terminación de la primaria, es importante hacer una comparación con el estudio realizado por el doctor Bosh et al, en donde reporta que las citopatías mitocondriales del tipo OPE Y KSS no presentan alteraciones intelectuales ni demencia1.
Los diagnósticos encontrados en el estudio fue con mayor frecuencia la OPE en 26.1%, KSS 17.4%, LHON en un 13%, MMGIE, enfermedad de Leigh, MELAS en 4.3% y sin diagnóstico un 30.4%. Los que solamente presentaron afección oftalmológica fueron un 43.5%, corresponden a los que presentaron diagnóstico de OPE y LHON. En cuanto la afección a otros órganos fue de 56.5% que son los pacientes con diagnóstico de enfermedad sistémica en los que encontramos (KSS, MMGIE, enfermedad Leigh, MELAS). Teniendo en cuenta que en 7 pacientes no se determinó un diagnóstico sindromático.
Las citopatías mitocondriales debidas a mutaciones en el genoma mitocondrial, tienen bien establecido un patrón de transmisión hereditaria por línea materna9. En este estudio encontramos que solo un 47.8% refirió presentar familiares con la misma patología, esto puede deberse a diversas causas: a falta de conocimiento de alteraciones en otros familiares, a casos con manifestaciones mínimas, a mutaciones de novo o a disfunciones mitocondriales secundarias a mutaciones en genes nucleares.
El antecedente de disminución de AV en la población estudiada se presentó en 69.6%, que correspondieron en su mayoría a LHON, OPE y un paciente que presentó retinosis pigmentosa sin diagnóstico definitivo. En varios pacientes se encontraron errores refractivos los cuales pudieron haber sido el síntoma principal, por lo que los pacientes acudieron con un facultativo y se les inició estudio completo. Los errores refractivos fueron en su mayoría la presencia de miopía de moderada a severa, teniendo en cuenta que la muestra es pequeña, se requerirían más pacientes para poder corroborar los datos observados y atribuirlos a una alteración del tipo mitocondrial.
Al momento de la exploración se tomó con mayor importancia la AV corregida, para tener una real agudeza visual sin la presencia de errores refractivos encontrándose que en 43.5% de los pacientes en el OD presentaron 20/20 y 47.8% en el Ol, una visión menor de 20/100 se encontró en 17.3% en ambos ojos. Estos resultados se identificaron en individuos cuya patología de base se asocia con mayor afección de globo ocular, en segmento anterior, retina y nervio óptico. Hubo concordancia entre afección de nervio óptico y pruebas cromáticas con afección importante en pacientes con diagnóstico de enfermedad de Leigh, LHON o MELAS.
La presión intraocular no se encontró alterada en ningún paciente.
La ptosis palpebral fue el signo inicial en la mayoría de los pacientes, ya sea bilateral o con afección primero a un ojo y meses o años después el segundo. En la exploración oftalmológica se encontró que la apertura palpebral menor de 10 mm, que se considera como ptosis se presentó en 56.5% en ambos ojos con una función del elevador de menor de 10 mm lo cual índica patología de 69.4% en OD y 65% Ol, con referencia a 30.4% que presentaron una nula función del elevador en ambos ojos, en consecuencia el fenómeno de Bell fue negativo en 56.5%
Los pacientes que presentaron diplopía como inicio del padecimiento solo fueron 8.7%; y solo en un caso se corroboró con exploración mediante lente rojo. Este paciente fue sometido a una cirugía de extracción de catarata con implante de lente intraocular del OD y fue posterior al procedimiento que refirió la presencia de diplopía.
En la exploración oftalmológica encontramos con frecuencia alteraciones en la movilidad ocular, llamó la atención que el 95.7% de los afectados refirieron no haberse dado cuenta que presentaron alteraciones en los movimientos oculares. Es posible que esto se deba a un limitado nivel educativo de los pacientes y sus familiares. El 52.2% de los casos de afección de músculos extraoculares cursó con oftalmoplejía completa. El reflejo optocinético se encontró negativo en 78.3% que es secundario a la afección muscular no a afección del sistema nervioso central, en la mayoría de los casos.
Se encontraron diferentes tipos de desviaciones oculares de las cuales en su mayoría fueron del tipo de exotropías simples 56.5%, exotropía con hipertropía en un paciente y exotropía con hipotropía en otro paciente, 34.8% se encontraron en ortoposición, teniendo en cuenta que a una paciente se le realizó corrección del estrabismo quirúrgicamente y no podemos saber el tipo de desviación que presentaba. Es relevante que no encontramos ninguna endotropia; proponemos que la mayor frecuencia de exotropia se debe a que el músculo recto interno es el que tiene mayor función, por lo tanto mayor requerimiento energético, siendo probable que este fuera el primer blanco de disfunción mitocondrial.
En la exploración en la lámpara de hendidura se observaron diferentes alteraciones de importancia, en cuanto a las alteraciones conjuntiva les que se observaron en 17.4% en OD y 13. % en Ol cursó con hiperemia importante, esto pudiera estar relacionado con datos de irritación por exposición secundaria a la falta de cierre palpebral en los pacientes a los cuales se le corrigió quirúrgicamente la ptosis palpebral en una o varias ocasiones. Las alteraciones encontradas en la cornea en las cuales encontramos leucomas cornéales y pannus inferior pudieran ser secundarias a queratitis por exposición al igual que las afecciones conjuntivales por falta de cierre palpebral; estas alteraciones no las podríamos relacionar con la enfermedad de base ya que en la mayoría de los pacientes ya habían sido sometidos a cirugía de corrección de ptosis y los pacientes que no habían sido operados no presentaron este tipo de alteraciones corneales.
En cuanto a las alteraciones en el iris se encontró que sólo un paciente presentó coriectopia al que se le realizó la cirugía de extracción de catarata con implante de lente intraocular y por consiguiente presentó trauma indiano quirúrgico. En cuanto a alteración de reflejos tanto consensúales como fotodinámicos se encontró con la misma justificación secundario a la cirugía previa de catarata.
No se observaron alteraciones en al ángulo iridocorneal en ningún paciente, por lo cual no presentaron alteraciones en las presiones intraoculares, esto es de importancia ya que es una zona del globo ocular la cual no presentó cambios, aunque tenemos que tener en cuenta que la muestra de población es pequeña.
Las alteraciones del cristalino que se encontraron fueron datos de suma importancia ya que la mayoría de los pacientes que presentaron mayor afección sistémica se encontraron alteraciones en cristalino como opacidades puntiformes difusas. Esto es muy relevante ya que la función del cristalino es mantenerse transparente para poder funcionar como lente refractivo del globo ocular, para que este órgano mantenga su transparencia es necesario que se realice una deshidratación constante en la cual intervienen una gran cantidad mecanismos como bombas de Na/K y bomba de Ca para eliminar el agua, por lo que se utiliza una gran cantidad de energía. La afección de la cadena respiratoria condiciona un aumento en la concentración de calcio, redegradación de proteínas en aminoácidos con el consecuente aumento de osmolaridad, que da lugar a un aumento del agua en el interior del cristalino y hace que se opacifiquen las fibras corticales. Asimismo, el cristalino es muy susceptible al daño por radicales libres, por esto se pudiera pensar que hay una relación entre la mayor afección sistémica y la presencia de opacidades cristalinianas, aunque la muestra de nuestros pacientes es pequeña pero es un dato de sumo interés para investigaciones futuras. Se observó un paciente que presentó una catarata de tipo subcapsular posterior bilateral y datos sugestivos de retinosis pigmentosa, sabemos que es frecuente la asociación de retinosis pigmentaria con este tipo de cataratas. Aunque no se sabe bien el mecanismo de producción se piensa en que las cataratas son secundarias a daño de tóxico producto de la degeneración retiniana5. Los pacientes que presentaron las opacidad es puntiformes difusas fueron los que tenían los diagnósticos de MMGIE, enfermedad Leigh, dos pacientes con OPE, tres con KSS y cuatro pacientes sin diagnóstico definitivo.
A todos los pacientes se les realizó estudio completo bajo dilatación pupilar para observar las características clínicas de la retina, se les revisó con lente de 4 espejos bajo anestesia tópica. Se encontró que en un 52.2% en ambos ojos presentaron papila de coloración anaranjada que se considera normal, 26 % en ambos ojos presentaron palidez papilar que se clasificó en, + ligeramente pálida–+ + + + , palidez máxima sin llegar a la atrofia, 17.4% presentaron atrofia completa en ambos ojos, este porcentaje de pacientes se correlacionan con los datos de alteración cromática lo que nos indica una gran afección a nervio óptico principalmente; se observaron vasos centrales en el 87% sin alteraciones, 13% presentaron cambios vasculares adelgazados en la periferia retiniana.
En las características clínicas de la retina encontramos cambios en la coloración de la retina que se interpreta como cambios en el epitelio pigmentado de la retina periférica en 39% OD, Ol 43.5%, cambios centrales en polo posterior en 26.1% OD, Ol 21.7% que en estos pacientes se encontraron cambios maculares de tipo degenerativos, tres pacientes presentaron cambios de tipo fondo coroideo que se correlaciona con los pacientes con errores refractivos del tipo de miopía leve–moderada y sin datos clínicos de alteración en el 17.4%.
En cuanto a los estudios para ver funcionamiento de fotorreceptores, células bipolares y de Müller que se observa con el electrorretinograma encontramos que hay una diferencia entre los datos clínicos encontrados y los datos fisiológicos, que fueron anormales en solo un 48.7% en comparación con los datos clínicos que se encontraron en mayor cantidad de pacientes. No hemos encontrado una explicación convincente, pero se pudiera pensar en la cantidad de afección por zonas de la retina y que las zonas observadas clínicamente son las que dan datos de afección, o de localización de las fibras alteradas. Son similares a los datos del estudio de la conducción de fibras del nervio óptico (potenciales visuales evocados), ya que en un 52.2% hubo anomalías, pero 43.5% (OD). 47.8% (Ol) de los pacientes presentaban una AV de 20/20, esto lo pudiéramos explicar ya que la sensibilidad del estudio a las fibras afectadas y se denominan anormales con un simple retraso en la conducción a nivel prequiasmático, estos son datos que se tienen que continuar en estudio para realizar una buena correlación clínica con estudios complementarios para beneficio de los pacientes.
Los campos visuales (CV) que se realizaron fueron de tipo Goldmann en donde se observó que un 56.5% de los pacientes presentaron CV normales, se observaron alteraciones de tipo hemianóptico, disminución concéntrica que estos se presentaron en las pacientes que tenían una mayor afección retiniana periférica, escotomas cecocentrales, isla de visión central y en dos pacientes no fueron valorables por la poca cooperación de los pacientes.
La electromiografía fue anormal en un 65.2%. explicable por la afección muscular en diferentes grados según el diagnóstico y grado de afección.
No hay un tratamiento disponible para este tipo de patologías pero es de suma importancia, dar un soporte sistémico en los pacientes que lo requieran como es el caso de los pacientes que presentan alteraciones cardiacas, es necesario la colocación de marcapasos, tratamiento médico para los pacientes con epilepsia, mejoramiento de la hipoacusia, realizar cirugía de corrección de ptosis en lo posible no dejando cierre inadecuado de la apertura palpebral y mantenimiento con lubricantes oculares tratando complicaciones que se presenten del tipo de queratitis por exposición, todo esto para mejorar la calidad de vida de los pacientes. El tratamiento metabólico incluye creatinina, coenzima Q, idebenone, succinato, menadion, riboflavina, nicotinamida, vitamina E, ácido ascórbico, tiamina y L– carnitina5–10.
Es indispensable iniciar tratamiento de las diferentes alteraciones sistemicas para brindar una mejor calidad de vida a los pacientes y disminuir las complicaciones.
CONCLUSIONES
Al finalizar el estudio se encontró que las diferentes citopatologías mitocondriales tienen ciertas características clínicas oftalmológicas similares, que según el grado de afección sistémica aumenta el grado de datos oftalmológicos.
En algunos pacientes que presentaron ptosis palpebral se les diagnóstico como miastenia gravis dándoles por largo tiempo tratamiento para esta patología incluso en algunos pacientes se les realiza timectomla sin presentar ninguna mejoría, por lo que es de suma importancia tener en cuenta este tipo de patologías realizar estudio completo de nuestros pacientes para dar un diagnóstico certero.
El estudio y observación de estos pacientes debe de ser multidisciplinario de las diferentes patologías para un buen seguimiento de los pacientes, pero siempre es indispensable tener y conocer este tipo de patologías par dar un buen apoyo, diagnóstico, evaluación de los pacientes.
Con las nuevas tecnologías se sabrá mejor en los próximos años, es indispensable tener los conocimientos más avanzados en este tipo de enfermedades, mantenerse al tanto de los nuevos descubrimientos, para el bienestar de los pacientes.
REFERENCIAS
1. Nissenkorn Anorem Mo, Zeharia Avraham MD. Neurologic presentations of mitocondrial disorders. J Chid neruol 2000;15:44–8. [ Links ]
2. Bosbach Simone, Kornublum Cornelia, Schooer Rolf, Wagner Michel. Exacutive and visuopatial deficits in patiens with chronic progressive external oftalmoplegia and Keams–Sayre syndrome. Brain 2003;126 (5)1231–40. [ Links ]
3. Martinez–Fernandez E, MD, Gil–Peralta phD. Mitochondrial diseases and Stroke. Stroke, 2001; 32:2507–10. [ Links ]
4. Luis Ruano Calderón. Artículo de revisión para entender las mitocondriopatías. Arch Neurocien Méx 2002; Vol. 7(4): 192–6 [ Links ]
5. Mirón Yanoff, Jay S Duker. Opthalmology. Mosby International Ltd 1999; 4.1.1–4.9.2 [ Links ]
6. Shanke, Yingyng Tang Sara. Identical mitochondrial DNA deletion in a woman with ocular myopathy and her son with Pearson syndrome. Am J Hum Genet 2002;.1:679–83. [ Links ]
7. Mehndiratta MM, P. Agarwal. Neurological mitochondrial cytopathies. Neuro India 2002; 50:162–67, [ Links ]
8. Ichlzo Nishino MD, Antonella Spinazzola. Mitochondrial neurogastrointestinal encephalomyopathy; autosomal recessive disorder due to thymidine phosphorylase mutation. Ann Neurol 2000;47:792–800. [ Links ]
9. Biousse Valerie, Newman Nancy J. Neuro–ophthalmology of mitochondrial diseases. Curr opin Neurol 2003;16:35–43, [ Links ]
10. Shoffner John M, MD. Mitochondrial myopathy diagnosis. Seminars in Neurology 1999; 19 (4)341 –51. [ Links ]

