











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La mucopolisacaridosis tipo II (MPS II) o síndrome de Hunter es una enfermedad multisistémica y progresiva con modelo de herencia recesiva ligada al cromosoma X, y está causada por variantes patogénicas en estado hemicigoto en el gen IDS, localizado en el brazo largo del cromosoma X, región 2 y sub-banda 8, la cual codifica para la enzima lisosomal iduronato-2-sulfatasa.1 El déficit de actividad de esta enzima es responsable del acúmulo tisular de los glucosaminoglucanos sulfato de dermatán y sulfato de heparán.2,3 Se han descrito más de 450 mutaciones, de las cuales 360 se han relacionado a la MPS II.4
En México, la incidencia estimada de MPS II es de 0.15 por 100,000 nacimientos.5 Por lo general, los casos se presentan en el sexo masculino.6 El cuadro clínico varía desde formas progresivas tempranas y lentamente progresivas, dependiendo del grado de afectación neurológica,1 siendo el fenotipo progresivo temprano el más frecuente, hasta en 60% de los casos.2
Al nacimiento no son evidentes las características clínicas.2 La edad de aparición de los datos clínicos oscila entre los 18 meses a los cuatro años en fenotipos progresivos tempranos, y dos años después en lentamente progresivas.7
Los pacientes presentan un amplio espectro de síntomas y signos clínicos, que incluyen características faciales por la infiltración de tejidos blandos (98%), hepatoesplenomegalia (88%), rigidez articular o disostosis múltiple (87%), macroglosia (84%), hernias (69%), obstrucción nasal (28%),8 además de engrosamiento de vías aéreas, síndrome de apnea obstructiva del sueño, hipoxemia crónica, talla baja, valvulopatía mitral y melanocitosis dérmica. En las formas graves aparecen trastornos del comportamiento y disminución progresiva del coeficiente intelectual. Se han reportado convulsiones tónico-clónicas, degeneración retiniana e hipoacusia. En las formas lentamente progresivas, los pacientes tienen inteligencia normal, características faciales poco finas, disostosis leve y supervivencia prolongada.2 Es poco frecuente la opacidad corneal.4
El reconocimiento del fenotipo del paciente permite el diagnóstico diferencial con otras enfermedades lisosomales.6 La elevación glucosaminoglucanos en orina tiene valor para el diagnóstico pero no lo confirma.4 La medición cualitativa determina el tipo de sulfato elevado y permite distinguir el tipo de mucopolisacaridosis.6 El diagnóstico requiere la demostración de la disminución de la actividad enzimática, lo cual se confirma al identificar una variante patogénica en el IDS.9 Las pruebas moleculares se recomiendan principalmente para el asesoramiento genético y la detección de portadores.2
Hasta el año 2006, el tratamiento había sido de soporte o paliativo. En ese año, en EUA se aprobó la infusión intravenosa de idursulfasa como terapia de reemplazo enzimático (TRE) para pacientes con MPS II. La idursulfasa es una enzima que hidroliza ésteres de 2-sulfato para proveer enzimas de manera exógena a lisosomas de varias estirpes celulares.10 La respuesta depende de la gravedad y de la edad de inicio del tratamiento.11
Existen reportes de casos de pacientes con MPS II en Latinoamérica, la mayoría sin tratamiento con TRE.4,12 El objetivo del presente reporte es describir el caso de un niño con MPS II, en quien se ha administrado idursulfasa como TRE.
Presentación del caso
Masculino de tres años al momento del diagnóstico. Sin antecedentes familiares de MPS II, tampoco de consanguinidad o endogamia. Producto del primer embarazo de madre de 27 años. El padre de 26 años, y una hermana de dos años aparentemente sana. El embarazo tuvo adecuado control prenatal. Presentó amenaza de aborto en el primer trimestre. Se detectó restricción del crecimiento intrauterino y preeclampsia a las 36 semanas de gestación. Fue obtenido por cesárea a las 36 semanas de gestación, sin requerir maniobras de reanimación avanzada. Peso al nacer 2,235 g (-2.6 DE), talla 47 cm (-1.5 DE), perímetro cefálico 33 cm (-1.2 DE), Apgar 9-10.
Desarrollo psicomotor: sonrisa social a los dos meses, sostén cefálico a los cuatro, sedestación a los siete y bipedestación a los 11 meses de edad. No gateó, y la deambulación comenzó a los 18 meses. La prueba de tamiz metabólico tuvo un resultado normal.
Desde el nacimiento se observó con respiración ruidosa; a los seis meses de edad con datos de obstrucción respiratoria y ronquido por las noches. A los dos años se hizo evidente en la cara la infiltración de tejidos blandos; además tenía el cabello crespo y puente nasal deprimido. Se refieren infecciones de vías respiratorias recurrentes. A los tres años, se percibe que había lenta progresión del habla, además dificultad en la memoria y atención, así como baja tolerancia a la frustración, y aún no había control de esfínteres. Acude por presentar caídas frecuentes, articulaciones rígidas con limitación de la movilidad y retraso en el lenguaje.
Al examen físico, los signos vitales eran normales. Peso 16.7 kg (> p85), talla 101 cm (p > 85) y PC 51 cm (p85). Cráneo dolicocéfalo, prominencia frontal, cabello crespo, cejas gruesas y pobladas, pupilas isocóricas, normorreflécticas, córnea y cristalino transparentes. Arcos supraorbitarios prominentes, puente nasal deprimido, narinas antevertidas con rinorrea hialina, filtrum largo, hipertrofia gingival, macroglosia, hipertrofia amigdalina grado IV. Pabellones auriculares gruesos de implantación baja (Figura 1). Tórax asimétrico con protrusión y desviación esternal a la izquierda, sin datos de dificultad respiratoria. Campos pulmonares con roncus leve. Precordio normal. Abdomen con esplenomegalia 5 a 6 cm bajo el reborde costal y hernia umbilical reductible. Extremidades con desviación radiocubital bilateral, arcos de movilidad limitados: codos con rezago de 30o a la extensión, subluxación de articulación escapulotorácica derecha, omoplato fuera de la línea axilar, hombro con limitación en la flexión bilateral de 140o, muñeca con limitación a la extensión en últimos 30o, dedos en semiflexión leve con limitación a la extensión (Figura 2). Miembros torácicos hipertónicos, con reflejos de estiramiento muscular disminuidos. Miembros pélvicos con fuerza cadera 2/5 rodilla 3/5, logra giros, signo de Gower positivo, rodilla con limitación en la extensión 20o, tobillos con limitación a la dorsiflexión más de 10o, acortamiento de tendón calcáneo. Mancha mongólica en región inguinal derecha de 15 cm.

Figura 1: Paciente a los tres años con facies sugerente de infiltración de tejidos blandos, cráneo dolicocéfalo, prominencia frontal, cabello crespo, cejas gruesas y pobladas, arcos supraorbitarios prominentes, puente nasal deprimido, narinas antevertidas, pabellones auriculares de implantación baja.
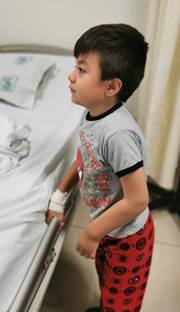
Figura 2: Paciente a los tres años, con facies sugerente de infiltración de tejidos blandos, implantación baja de pabellones auriculares, rezago de 30o a la extensión de codos.
La biometría hemática, química sanguínea, electrolitos séricos, pruebas de función hepática, pruebas de función tiroidea y el examen general de orina fueron normales, a excepción de fosfatasa alcalina de 374 U/L (normal: < 130 U/L).
En ultrasonido (US) abdominal se detectó esplenomegalia: bazo de 85 mm con ecogenicidad y forma conservada. Hígado con tamaño en el límite superior máximo normal. Radiografía de manos con dorsiflexión de articulaciones interfalángicas proximales y distales (Figura 3). Ecocardiograma: situs solitus en levocardia, sin patología valvular. Umbral auditivo disminuido (60 dB (VN 0-25 dB)). Evaluación psicológica: trastorno del desarrollo psicomotor, trastorno por déficit de atención clínico y disfasia del desarrollo.
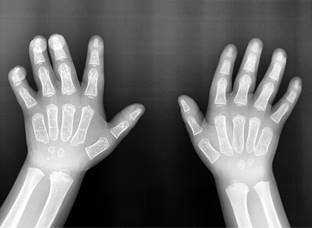
Figura 3: Radiografía anteroposterior de manos que muestra dorsiflexión de articulaciones interfalángicas proximales y distales.
Tamiz metabólico ampliado de 69 parámetros con resultado sospechoso para MPS I y II. La actividad enzimática de alfa-L-iduronidasa fue normal (5.88 nmol/mL (normal: 2.02-16.1 nmol/mL)), por lo que se descartó MPS I. Pero los niveles de iduronato sulfatasa se encontraron disminuidos (< 2.8 umol/L/h (normal > 5.6 umol/L/h)). En el estudio molecular del gen IDS se identificó variante patogénica en c.14036>A p.(Arg468Gln) en el exón 9. La madre resultó portadora de la misma variante.
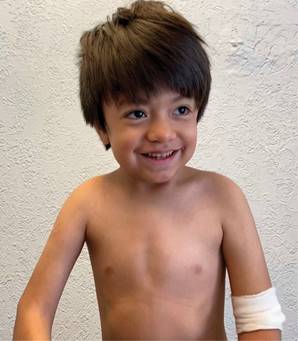
Con los resultados anteriores, se decidió iniciar administración de idursulfasa intravenosa 6 mg una vez por semana a dosis de 0.5 mg/kg, además de terapia del lenguaje y física con movilización asistida de arcos de movimiento articulares. Durante los 24 meses que ha tenido de tratamiento se ha monitorizado la evolución de variables antropométricas, volúmenes del hígado y el bazo por US, así como mediciones del rango de movimiento articular. En la última evaluación, a la edad de cinco años, el peso era de 23.2 kg (percentil 95), talla 117 cm (percentil 95) y perímetro cefálico de 53 cm (percentil 85) (Figura 4A y B). También se observó que en la cara había menor infiltración de tejidos blandos (Figura 5). Por US, el hígado tenía longitud de 114 mm, y el bazo medía 70 mm. Asimismo se registró la mejoría de la movilidad articular: disminución de 10o de rezago a la extensión en articulación de codos, muñecas, tobillos y rodillas, además de menor limitación a la extensión de articulaciones interfalángicas distales. Durante este periodo no tuvo infecciones de vías aéreas, hospitalizaciones ni reacciones adversas al tratamiento. Sin embargo, el paciente continúa con trastorno del desarrollo y del comportamiento, principalmente trastorno por déficit de atención e hiperactividad, además de apnea del sueño. Se programó para adenoamigdalectomía, pero fue suspendida por dificultad para la intubación orotraqueal.
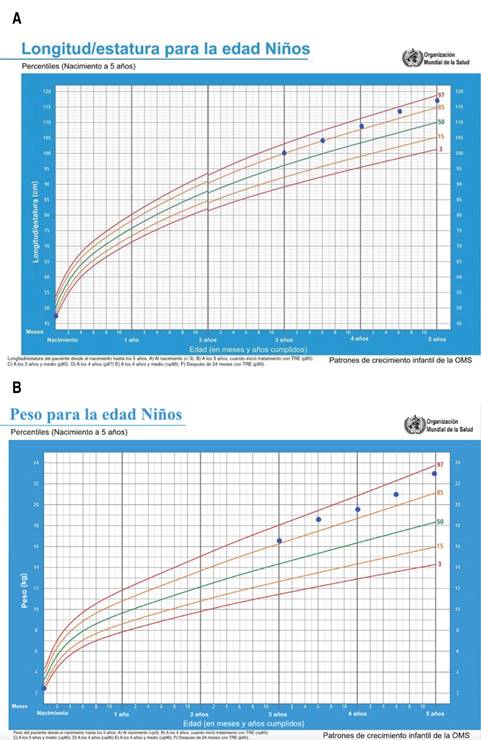
Figura 4: A) Longitud/estatura del paciente desde el nacimiento hasta los cinco años. a) Al nacimiento se encontraba en percentil 3 (p3) b) A los tres años, cuando inició tratamiento con terapia de reemplazo enzimático (p85). c) A los tres años y medio (p85). d) A los cuatro años (p87). e) A los cuatro años y medio (> p90). f) Después de 24 meses con terapia de reemplazo enzimático (p95). B) Peso del paciente desde el nacimiento hasta los cinco años. a) Al nacimiento (<p3). b) A los tres años, cuando inició tratamiento con terapia de reemplazo enzimático (>p85). c) A los tres años y medio (>p85). d) A los cuatro años (>p85). e) A los cuatro años y medio (>p90). f) Después de 24 meses con terapia de reemplazo enzimático (p95).
Discusión
El presente caso muestra los datos clínicos más frecuentes de la MPS II, los cuales son característicos del fenotipo progresivo temprano de la enfermedad, destacando el retraso psicomotor, la limitación de la movilidad articular y la afección respiratoria. Mientras que la confirmación del diagnóstico fue por niveles bajos de actividad enzimática de iduronato sulfatasa y por la identificación de la variante patogénica en c. 1403G>A p.(Arg468Gln) en el exón 9 del gen IDS.3,13 Esta variante patogénica se relacionó con MPS II por Whitley y colaboradores.14 Y, con el propósito de disponer de un panorama más completo esta misma variante, se buscó en abuela y tías maternas, pero los resultados fueron negativos. Así, debido a que esta variante (o mutación) sólo fue identificada en la madre, entonces se consideró de novo, lo cual ocurre en 20% de los pacientes.3 Esta variante se ha observado tanto en el fenotipo progresivo temprano, como en el fenotipo lentamente progresivo por lo que no parece haber correlación del genotipo con la expresión clínica.13
Asimismo, señalamos que, como ya se ha reportado previamente, nuestro paciente ha cursado con afección de su capacidad cognitiva, y problemas de comportamiento (hiperactividad), lo cual no se modifica con la TRE. Además, parece necesario evaluar la función auditiva, ya que la hipoacusia puede agravar los trastornos del comportamiento y la dificultad en el aprendizaje.7
Por otro lado, las infecciones de vías aéreas superiores recurrentes son causa de morbilidad en MPS II.8 La sintomatología respiratoria es producida por depósito de los glucosaminoglucanos en tejido conjuntivo de región orofacial y en la vía aérea, incluyendo deformidades en tórax.8 Como consecuencia, se ha descrito que estos casos tienen respiración ruidosa exacerbada en la noche, problema que aumenta con la edad. También pueden padecer síndrome de apnea obstructiva del sueño hasta en 69%, por lo que se recomienda realizar pruebas de función pulmonar y polisomnografía.15 Mientras que, a largo plazo desarrollan cor pulmonale.10 La adenoamigdalectomía puede ayudar a reducir la obstrucción; sin embargo, como ocurrió en nuestro caso, es común la dificultad a la intubación endotraqueal.7
La forma recombinante de iduronato-2-sulfatasa humana ha sido aprobada en más de 50 países para la TRE del síndrome de Hunter. La dosis recomendada de idursulfasa es de 0,5 mg/kg de peso, administrada IV una vez a la semana, en infusión de una a tres horas.16 Pero se debe considerar que la decisión de usar este tratamiento, en general, se ha basado en un solo ensayo clínico controlado realizado en niños mayores de cinco años, donde se determinó mejoría en prueba de la marcha, aumento de capacidad vital forzada, disminución del volumen del hígado y el bazo, así como de los niveles de glucosaminoglucanos en orina.10,17 Sin embargo, idursulfasa no modifica los síntomas neurológicos, ya que no cruza la barrera hematoencefálica.11
Este tratamiento también se ha otorgado en niños menores de cinco años, y aunque la experiencia es limitada,18 es probable que el resultado sea mejor entre menor edad de inicio de la TRE.8,19,20 Aunque estudios recientes a largo plazo indican que, cuando ya se han establecido complicaciones, la TRE no tiene beneficio, como en problemas cardiacos.21

