











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Guillain-Barré syndrome (GBS) is the most frequent cause of acute flaccid weakness worldwide, with an incidence of 1-2 cases/100,000 person-years1. GBS is classically considered an immune-mediated, ascending, symmetric polyradiculoneuropathy usually preceded by an infection that may occur at any age. However, its incidence and severity increase with age, generally associated with axonal damage, less involvement of cranial nerves, and slower functional recovery2. GBS represents a neurological emergency as, despite proper treatment, up to 20% of patients will become seriously disabled, and approximately 5% will die1. Regardless of recent advances in GBS knowledge, mortality in Mexico has been reported to be as high as 12%3,4.
While vaccines have been historically linked to GBS, especially seasonal influenza vaccines, epidemiological studies indicate that they do not increase GBS incidence5. However, vaccination campaigns against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have raised questions of whether these newly-developed vaccines approved under emergency conditions may increase the risk of developing GBS6,7. This review discusses the current understanding in pathophysiology, clinical presentation, diagnostic criteria, and management of GBS, focusing on recent Mexican data, including the evidence of potential associations between SARS-CoV-2, anti-SARS-CoV-2 vaccines, and GBS.
EPIDEMIOLOGY AND POTENTIAL TRIGGERS
The overall incidence of GBS is higher in men than in women (0.86 vs. 0.57 cases/100,000 person-years)1. GBS epidemiology varies regionally. In Asian countries, incidence ranges from 0.44 to 3.25 cases/100,000 person-years, with acute motor axonal neuropathy (AMAN) being the predominant electrophysiological variant. In Europe, incidence ranges from 0.94 to 1.91 cases/100,000 person-years, and the predominant electrophysiological variant is acute inflammatory demyelinating polyneuropathy (AIDP). On the other hand, its epidemiology has marked variability in the Americas. For example, the United States reports 2.2 cases/100,000 person-years with AIDP predominance, while in Central and South America, the reported incidence ranges from 0.4 to 7.63/100,000 person-years1.
In 2010, Mexico reported 4 cases/100,000 person-years of acute flaccid weakness, and by 2019, a much lower incidence of 0.71 cases/100,000 person-years8. These differences between the Americas and the rest of the world may be related to differences in operational definitions and because in most Latin American countries, GBS reports mostly depend on passive epidemiological surveillance systems. The most common electrophysiological variant in Mexico is AMAN, probably associated with the increased incidence of Campylobacter jejuni gastrointestinal infections9,10; despite this association, in Mexico, GBS has a seasonal distribution with a peak of axonal variants during the summer, while the AIDP variant is more frequent in winter, possibly associated to a higher incidence of respiratory infections11.
About two-thirds of cases have a preceding infection 3-6 weeks before symptom onset. In case–control studies, C. jejuni, Mycoplasma pneumoniae, Haemophilus influenzae, Cytomegalovirus, Epstein-Barr virus, Hepatitis E virus, and Influenza A virus have been temporally associated with its development10. In addition, reports have demonstrated an increase in cases during outbreaks of specific pathogens such as Zika and Chikungunya. Interestingly, an association between Chikungunya, Zika, and GBS is unclear in Mexico12.
CORONAVIRUS DISEASE 2019 (COVID-19) AND IMMUNIZATION AGAINST SARS-CoV-2
Throughout the COVID-19 pandemic, a link between GBS and SARS-CoV-2 remains controversial. A population-based epidemiological study from the United Kingdom reported that the surge of COVID-19 cases during the first pandemic wave did not correlate with an increase in the expected incidence of GBS compared to pre-pandemic records; nevertheless, this finding might be related to the lockdown measures imposed by the pandemic, reducing the transmission of GBS-inducing pathogens13. Still, several studies have reported GBS cases after SARS-CoV-2 infection, suggesting a parainfectious mechanism14,15. However, a causal relationship is yet being debated10.
Vaccines have been historically linked to GBS; however, the last epidemiological association occurred almost five decades ago in association with the H1N1 influenza vaccine, when the risk for GBS increased 7.3-fold among immunized people5. No other vaccines have been directly linked to GBS ever since. As for the SARS-CoV-2 vaccines, some reports suggest a lack of causal association between the currently available mRNA-based (BNT162b2 and mRNA-1273) vaccines and GBS6,7,16.
A recent Mexican study conducted among 3,890,250 recipients of the BNT162b2 vaccine reported an observed incidence of 0.18/100,000 administered doses, suggesting that GBS among recipients of this vaccine may occur at the expected community-based rate7; however, current incidence among the unvaccinated population against COVID- 19 is still undetermined; therefore, these preliminary results should be taken with caution. On the other hand, epidemiological associations with the adenovirus-vectored Ad26.COV2.S (Janssen) and AZD1222 (AstraZeneca) anti-SARS-CoV-2 vaccines with GBS have been made, posing a red flag nonetheless6,17.
It has been hypothesized that contaminating proteins or other vaccine components may elicit anti-ganglioside antibody production18,19; still, these potential mechanistic associations remain to be elucidated. There are no current recommendations on subsequent immunization for two-dose regimen vaccines or boosters after developing GBS as an adverse event. Decisions must be taken on a case-by-case basis; in patients with non-SARS-CoV-2 vaccine-related GBS, it has been suggested that subsequent immunizations should be administered no less than 3 months after the last dose18. In the case of anti-SARS-CoV-2 vaccines, we suggest using another vaccine different from the initial one if available.
PATHOPHYSIOLOGY
Historically, GBS was thought to be a single-mechanism disease affecting the peripheral nerves. Further electrodiagnostic testing and pathological analysis have proved that at least two mechanisms are involved, resulting in demyelinating and axonal injury. Despite the discovery of anti-ganglioside antibodies, which opened the pathway toward identifying specific antibodies directed against the neuronal axon10,20, its exact cause and pathogenesis remain incompletely understood.
Gangliosides are glycolipids with one or more sialic acids, as n-acetylneuraminic acid (NANA) located within the cell surface that oversees cellular recognition and communication between cells. The number of NANAs determines the anti-ganglioside antibodies subtype: one = mono (GM), two = di (GD), three = T (GT), and four = Quattro (GQ)20. To date, there are no specific anti-ganglioside antibodies found in GBS demyelinating variants; those antibodies are only observed in axonal variants, as well as in Miller-Fisher syndrome (MFS) and Bickerstaff brainstem encephalitis (BBE) (Table 1). Some myelin proteins and neurofascin antibodies are currently being studied for AIDP. The anti-ganglioside antibodies found in AMAN and acute motor and sensory axonal neuropathy (AMSAN) electrophysiological variants are GM1 and GD1a, while GQ1b and GT1b are observed in MFS, BBE, and pharyngeal-cervical-brachial variants of GBS. GM1 and GD1a are found in patients who developed GBS after C. jejuni infection20.
Table 1. Guillain-Barré syndrome, Miller-Fisher syndrome and Bickerstaff brainstem encephalitis spectrum variants, clinical features, and associated anti-ganglioside antibodies
| Variant | Frequency | Clinical features | Possible cranial nerve involvement | Associated antibodies | Electrophysiological variant |
|---|---|---|---|---|---|
| Guillain-Barré syndrome spectrum variants | |||||
| Classic GBS | 30-90% | Classic progressive symmetrical weakness with/without sensory signs and areflexia, usually with dysautonomia | Yes | Unknown | AIDP |
| Pure motor variant | 5-70% | Progressive motor weakness without sensory signs | Yes | GM1, GD1a | AMAN |
| Paraparetic variant | 5-10% | Progressive weakness restricted to the legs | No | GM1, GD1b | Axonal |
| Pure sensory variant | < 1% | Progressive sensory symptoms without weakness | No | GD1b | N/A |
| Pharyngeal-cervical-brachial variant | < 5% | Progressive weakness restricted to pharyngeal, cervical, and brachial muscles | Yes | GT1a, GQ1b | Equivocal |
| Bilateral facial palsy with paresthesia variant | < 5% | Progressive bilateral facial palsy, with limb paresthesia | Yes | Unknown | AIDP |
| Acute pharyngeal variant | < 1% | Progressive acute weakness restricted to pharyngeal muscles | Yes | GT1a | Equivocal |
| Miller-Fisher syndrome spectrum variants | |||||
| Classic Miller-Fisher variant | 4-25% | Progressive ophthalmoparesis, ataxia and areflexia | Yes | GQ1b, GT1a | Normal |
| Acute ophthalmoparesis | < 1% | Progressive ophthalmoparesis | Yes | GQ1b | Normal |
| Acute ataxic neuropathy | < 5% | Progressive acute ataxia | No | GM1 | Axonal |
| Acute ptosis | < 1% | Progressive acute ptosis | Yes (only ptosis) | GQ1b | Normal |
| Acute mydriasis | < 1% | Progressive acute mydriasis | Yes (only mydriasis) | GQ1b | Normal |
| Bickerstaff brainstem encephalitis spectrum variants | |||||
| Classic Bickerstaff brainstem encephalitis | < 5% | Progressive ophthalmoparesis, ataxia, areflexia, pyramidal signs and hypersomnolence | Yes | GQ1b, GT1a | Axonal/Normal |
| Acute ataxic hypersomnolence | < 1% | Progressive ataxia and hypersomnolence | No | GQ1b | Normal |
| Overlap syndromes | Unknown | Overlap between GBS/MFS/BBE clinical syndromes | Yes | GM1, GT1a, GQ1b | AIDP/Axonal/Normal |
GBS: Guillain-Barré syndrome; MFS: Miller-Fisher syndrome; BBE: Bickerstaff brainstem encephalitis; AIDP: acute inflammatory demyelinating polyneuropathy; AMAN: acute motor axonal neuropathy. Modified from Shahrizaila N, Lehmann HC, Kuwabara S (2021)20.
The most accepted mechanistic hypothesis for GBS development is molecular mimicry producing cross-reactivity between bacterial lipo-oligosaccharides and neuronal gangliosides. Peripheral nerve injury in GBS is mediated by T cells, macrophages, and complement activation. Patients with demyelinating features usually recover. On the other hand, the recovery for axonal variants is generally slower than in demyelinating forms, and it depends on the degree of nerve injury20,21. Some patients with axonal variants may have a quick recovery explained by a functional conduction block without axonal injury. The former may occur during the initial stages; however, if an axonal injury is already present, recovery depends on the extent of damage22.
CLINICAL VARIANTS AND FEATURES
GBS is a heterogeneous disorder with several clinical variants. GBS presents in three stages; the first (progressive) is characterized by a monophasic, progressive course of symptoms, usually lasting 2-4 weeks; in the second (plateau) phase, symptoms gradually decrease but may persist from weeks to months. In the third (recovery) phase, symptoms gradually improve. Two-thirds of patients will have a preceding acute or subacute infection within 3-6 weeks before symptom onset10,20.
The classical GBS clinical variant (sensorimotor) is the most common, occurring in 55% of cases (Table 1 and Fig. 1). It usually presents with acute weakness and sensory alteration of all limbs, cranial nerve involvement, and autonomic dysfunction. In most instances, the weakness starts in the lower limbs, ascending to the upper limbs within several days or weeks -generally < 4- (Fig. 1). By the time weakness reaches the knees, it can also be detected in the upper limbs18,23. Overall, up to 38% of patients may develop at least one feature of autonomic dysfunction, most common among those with demyelinating forms. Ileus (42%), hypertension (39%), hypotension (37%), fever (28%), tachycardia or bradycardia (27%), urinary retention (24%), pupillary dysfunction (14.1%), or loss of sweating (9.9%) are the most common signs of autonomic instability24,25.
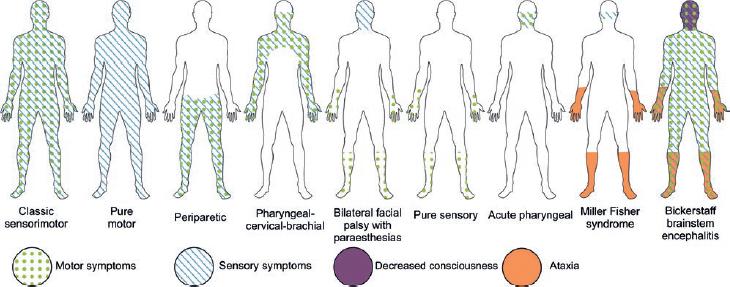
Other clinical variants of GBS include: pure motor, pure sensory, paraparetic, pharyngeal-cervical-brachial, acute pharyngeal variant, MFS, and BBE. Sometimes a variant may share characteristics with other variants (e.g., classic GBS with MFS features). These mixed variants are known as GBS overlap variants23,26. Usually, the pure motor, pharyngeal-cervical-brachial, and paraparetic variants have a more severe presentation, with gait loss and bulbar cranial nerve involvement leading to respiratory failure. Severe autonomic dysfunction is mostly seen in patients who develop severe weakness and respiratory failure20,23.
Despite having clinical manifestations different from the classical syndrome, MFS and BBE are included within GBS because they share similar pathophysiological characteristics. Within MFS, a triad of ophthalmoparesis, ataxia, and areflexia, we can find incomplete forms, such as the acute ophthalmoplegia variant, acute ataxic neuropathy, acute ptosis, and acute mydriasis20,23. In 50% of cases, MFS presents with overlapping BBE and classic GBS26. BBE clinically presents with progressive ophthalmoparesis, ataxia, areflexia, upper motor neuron signs, and hypersomnolence, sharing the presence of anti-GQ1b antibodies with the MFS. Interestingly, in BBE, there is a high expression of GQ1b antibodies in oculomotor nerves, neuromuscular spindles, and reticular formation20.
Cranial nerve involvement can be detected in up to 35% of cases, more frequently in the classic (sensorimotor) GBS variant. Bilateral facial palsy is the most common cranial nerve neuropathy (seen in 50% of patients presenting cranial nerve dysfunction). Ocular motor nerves involvement is more commonly associated with MFS, while the IX and X cranial nerves with the acute pharyngeal and pharyngeal-cervical-brachial variants20,23. Radicular and muscle pain are also frequent initial signs. In some cases, reflexes may be normal during the early stages of the disease, especially in axonal variants, and in some sporadic cases, hyperreflexia can be seen. However, as the disease advances, reflexes are progressively abolished. Twenty-five percent of patients will require invasive mechanical ventilation18,23. Forme fruste GBS occurs when patients exhibit incomplete clinical variants such as paraparesis restricted to the legs without clinical repercussion in the arms20.
DIAGNOSTIC CONSIDERATIONS
GBS is primarily a clinical diagnosis, although cerebrospinal fluid (CSF) analysis and nerve conduction studies (NCS) should always be performed. The US National Institute of Neurological and Communicative Disorders and Stroke (NINDS) published the first GBS diagnostic criteria precipitated in part by the increasing number of cases among recipients of swine flu vaccine in 1977, allowing an early diagnosis supported by clinical, laboratory, and electrodiagnostic features23. However, in 2011, the Brighton collaboration group developed updated case definitions for GBS and MFS occurring as adverse events following immunization, classifying patients according to the level of certainty depending on CSF analysis and NCS results into four levels, with level 1 representing the highest level of diagnostic certainty and 4 the lowest one20.
On CSF analysis, most patients may have elevated protein levels, but it may not be evident until 3 weeks after symptom onset. Therefore, the diagnosis may only rely on clinical presentation and electrophysiological testing during the initial stages. Nonetheless, performing a lumbar puncture is recommended in all patients to detect red flags despite an early presentation. CSF cell count is usually low (≤ 5 cells/µL); however, in up to 15% of cases, cell counts may range from 5 to 50 cells/µL. Pleocytosis greater than 50 cells/µL should raise suspicion for infectious or neoplastic origins (e.g., cytomegalovirus infection, Lyme disease, human immunodeficiency virus infection, sarcoidosis, or carcinomatous meningitis, among others)10,27.
All GBS patients should undergo electrophysiologic testing, not necessarily to confirm the diagnosis but to determine the nerve injury mechanism and prognosis. Timing of NCS is crucial as early studies may be normal or show very few signs of demyelination18,27. Pitfalls of electrophysiologic testing and diagnostic criteria will be addressed later in this review. Finally, we recommend testing all patients for serum anti-ganglioside antibodies to support the diagnosis and identify the disease immunophenotype. Anti-ganglioside antibody panels should include immunoglobulin G (IgG) and IgM for GM1, GM2, GD1a, GD1b, and GQ1b.
ELECTROPHYSIOLOGICAL VARIANTS AND CRITERIA
Electrophysiological studies are essential to define whether the injury mechanism is demyelinating or axonal. The demyelinating pattern is characterized by reduced nerve conduction velocity and prolonged latencies. On the other hand, the axonal pattern is characterized by reduced amplitudes, decreased compound motor action potentials (PAMC), conduction block, or an isolated prolonged latency or absence of F-waves28. Sural nerve-sparing is a key finding in GBS patients in the proper clinical setting21. The axonal variant has two subtypes: if involvement is limited to motor nerves, the variant is known as AMAN; if both motor and sensory nerves are involved, as ASMAN18. Table 2 shows the differences between axonal and demyelinating variants among the Mexican population.
Table 2. Clinical and paraclinical differences between axonal and demyelinating variants of Guillain-Barré syndrome in Mexican population
| Axonal | Demyelinating | |
|---|---|---|
| Overall frequency | 31-45% | 29-41% |
| Preceding diarrhea* | 35-47% | 24-44% |
| Preceding respiratory tract infection | 25-33% | 26-45% |
| Severe disease (GDS ≥ 3 at admission) | 85% | 83% |
| Cranial nerve involvement | 48% | 57% |
| Facial nerve involvement | 41% | 57% |
| Bulbar cranial nerve involvement | 27% | 30% |
| Sensory symptoms | 42-63% | 58-76% |
| Autonomic dysfunction | 25-31% | 15-28% |
| CSF cytoalbuminologic dissociation | 41-76% | 72-88% |
| Campylobacter jejuni positive testing | 44% | 37% |
| IMV requirement | 20-30% | 12-33% |
| Good functional prognosis** | 37% | 49% |
*Diarrhea < 4 weeks before symptoms onset.
**Able to walk 10 m independently at 3 months (GDS ≤ 2).
GDS: Guillain-Barré Disability Scale; IMV: invasive mechanical ventilation; CSF: cerebrospinal fluid.
Adapted from López-Hernández JC, et al. (2021)9.
The clinical and electrophysiological features of the various presentations can be seen in table 1. Interestingly, in MFS and BBE, electrophysiological studies are generally normal. However, acute ataxic neuropathy and classic BBE may have prolonged distal compound muscle action potentials (CMAP) in motor nerves without fulfilling axonal pattern criteria26. Some studies report that the most frequent electrophysiological variant is AMAN, which presents some clinical and paraclinical differences compared to the AIDP variant (Table 2)9,20. Although AMAN is the most prevalent variant in Mexico, only 35% of cases have preceding diarrhea, while respiratory tract infections precede in 25%. On the other hand, AIDP is preceded by diarrhea in 44.4% of cases or a respiratory tract infection in 26%9. AIDP is usually less severe and carries a better prognosis than AMAN, which has a faster course associated with an increased risk for invasive mechanical ventilation requirement10.
The first electrophysiological criteria for GBS were published by Hadden et al. in 1998; however, the cutoff values they proposed had low specificity for the demyelinating variant, particularly for latencies and nerve conduction velocities28. Furthermore, studies with an axonal pattern in subsequent studies demonstrated a demyelinating pattern; therefore, a single NCS was not enough to make the diagnosis. In addition, in up to 38% of patients, the electrophysiologic classification may change in follow-up studies29. The aforementioned caveats led various authors to question these criteria and propose new cutoff values to determine the nerve injury mechanism in early NCS studies.
Rajabally et al., in 2014, proposed new criteria that allowed for an early (< 7 days) and more accurate determination of the electrophysiological findings’ nature29, obtaining results similar to those published by Hadden et al. when the study is conducted between 3 and 10 weeks after symptoms onset. Furthermore, they demonstrated a reduction in equivocal variants. In 2017, Uncini et al. proposed new electrophysiologic criteria, suggesting that dynamic changes occur in peripheral nerves, and some cases may benefit from serial testing30.
For those reasons, we prefer using Rajabally’s or Uncini’s criteria to define the electrophysiological subtype with a single study. Within 3 to 8 weeks from disease onset, a second study is recommended in cases where the first study shows no clear demyelinating features, has low amplitude distal CMAP, or conduction block without temporal dispersion28. In addition to knowing the different criteria, the adequate performance of NCS should rely on an experienced electrophysiologist.
THERAPEUTIC CONSIDERATIONS
The first step in GBS treatment is to know which patients may benefit from immunotherapy or supportive treatment alone. The Guillain-Barré Disability Scale (GDS) allows clinical stratification of patients. This scale ranges from 0 to 6 and primarily assesses the gait and mechanical ventilation requirement23. However, as it only measures lower limb strength indirectly by evaluating gait, new scores should be sought to improve treatment selection.
Immunotherapy should be considered in patients unable to walk more than 10 m independently (GDS ≥ 3), those with severe autonomic dysfunction, rapid progression of weakness, bulbar muscles involvement, or respiratory failure within 4 weeks after symptoms onset. Treatment of mildly affected patients (GDS 1 or 2) is controversial as to whether it is cost- and risk-effective, as this subset of patients usually recovers faster due to a milder course. Patients with mild GBS should be observed and treated with immunotherapy only if severe clinical worsening occurs, for instance, if the patient is unable to walk unaided (GDS ≥ 3) due to rapidly progressive weakness, develops severe autonomic dysfunction, involvement of bulbar muscles, or respiratory failure18,20,27. MFS is considered a disease with a benign course; therefore, only supportive treatment is recommended unless an overlap variant is diagnosed or if the patient presents any of the previously listed features. In BBE, clinical severity always justifies acute treatment with intravenous Ig (IVIg) or plasma exchange (PLEX)26.
Acute treatment options include IVIg and PLEX. IVIg is recommended at a total dose of 2 g/kg administered during 5 consecutive days; as for PLEX, five sessions on alternate days (total volume exchange of 200-250 mL/kg or 40-50 mL/kg each session) are the current recommendation. Both treatments are equally effective as they accelerate recovery. IVIg started within 2 weeks after symptoms onset hastens recovery as much as PLEX, has a similar rate of adverse events, and is likelier to be completed18,20,27.
PLEX has been demonstrated to be effective when started within 4 weeks after symptoms onset10. Combining or switching therapy (PLEX followed by IVIg or vice versa) is not recommended as evidence is not clear on whether patients may benefit or not from switching therapies. Use of PLEX after IVIg is discouraged as PLEX may wash out IVIg31. Small volume PLEX (total exchange of 140 mL/kg over 8 days) is a safe and feasible treatment in low-income countries where IVIg and PLEX are unavailable or unaffordable10. In Mexico, the decision between IVIg and PLEX depends on treatment availability, and recent studies report that only 63-75% of patients are so treated3,4,9.
A trial re-evaluating the efficacy and safety of this drug among patients with severe GBS is ongoing (ClinicalTrials.gov Identifier: NCT04752566). Corticosteroids are not recommended for acute GBS as they do not improve short- or long-term outcomes, as demonstrated in several clinical trials27. Patients should be considered for intensive care unit admission if they develop progressive respiratory distress, bulbar muscles involvement, rapidly progressive weakness, an Erasmus GBS Respiratory Insufficiency Score (EGRIS) > 4, or severe autonomic dysfunction20,27.
Treatment-related fluctuations are characterized by clinical deterioration after initial improvement or stabilization within the first 8 weeks following treatment initiation, defined as a decrease of 1 or more points in the GDS. These can occur in 8-16% of patients, and the specific mechanism remains unclear. This subset of patients might benefit from receiving a second course of IVIg or PLEX20,27; however, further studies addressing this question are still needed. The benefits of a second course of IVIg were studied in the SID-GBS trial, which only included patients with a poor prognosis (score of ≥ 6) according to the modified Erasmus GBS Outcome Score (mEGOS), failing to demonstrate that patients with a poor prognosis benefit from a second IVIg course33. Even so, since that study only included patients with a severe course of the disease, further clinical trials are still needed to determine accurately if a subset of patients may benefit from this treatment strategy. Hence, with the available evidence, administering a second course of IVIg should be limited to research settings.
Supportive treatment must include continuous respiratory assessment, nasogastric tube placement if bulbar weakness is present to prevent aspiration pneumonia, management of autonomic dysfunction, early rehabilitation evaluation, delirium management, pain relief, and deep venous thrombosis prophylaxis. These conditions are essential to consider as they are the most common causes of death during the recovery phase20,27. In addition, pain management is fundamental since 89% of patients will develop this complication. Different kinds of pain have been reported, including radicular pain, paresthesia, muscle pain, visceral pain, and meningism. Gabapentin, pregabalin, and carbamazepine are recommended for neuropathic pain as these have proven to be effective for long-term management34.
Management and monitoring of autonomic dysfunction are imperative, as dysautonomia may develop in up to 70% of patients, remaining as a potentially deadly complication to keep in mind. Tachycardia is the most common sign of dysautonomia in 25-38% of patients. For sinus tachycardia, only monitorization is recommended as sinus blockers may cause fatal bradyarrhythmias. Non-pharmacological bradyarrhythmias and conduction blocks are infrequent but may require atropine administration or pacemaker implantation in some instances25.
Hypertension develops in 27% of patients. Therefore, for mild to moderate episodes of hypertension, only close monitoring of blood pressure is recommended and for severe hypertension, defined as a mean arterial pressure > 125 mmHg, treatment with either IV labetalol, esmolol, or nitroprusside is justified35. Patients presenting with fluctuations between hypertension and hypotension should be admitted to the intensive care unit, as antihypertensives may cause severe hypotension leading to circulatory collapse, and vasoactive agents can precipitate a hypertensive crisis. In that setting, the start-low, go-slow approach is recommended. Cardiovascular monitorization is vital in patients treated with PLEX, as it can lead to severe hypotension. Strength, gait, and swallowing rehabilitation provided by a specialist are mandatory in treating severe GBS cases25.
PROGNOSIS
EGRIS and mEGOS are helpful tools to predict respiratory and functional prognosis during the acute phase, respectively. EGRIS estimates the risk of respiratory failure within the first week of admission, considering the days between symptom onset and admission and facial/bulbar weakness. The total MRC sum score and the mEGOS performed on the seventh day following hospital admission predict the probability of being unable to walk independently within the first 6 months of follow-up; this score considers patient age, history of diarrhea within the past 4 weeks before symptoms onset, and the MRC sum score20,27.
Despite timely and proper treatment, up to 20% of patients will be unable to walk unaided at 6 months; however, some may still show functional improvement 3 to 6 years after the event. Risk factors for a poor functional outcome (inability to walk unaided) include older age, severe presentation, and mechanical ventilation requirement18. A recent Mexican study reported that patients aged >70 years had a delayed gait recovery compared to younger patients4. Mechanical ventilation directly impacts the functional outcome and prognosis. Rapid motor symptom progression and early cranial nerve involvement are predictors for mechanical ventilation and aspiration risk3.
GBS is still a life-threatening illness. The reported mortality rate in Mexico of 10-12% is slightly higher than that described in other parts of the world3,4,9. Before IVIg and PLEX, GBS had a reported mortality rate ranging from 3-13% worldwide, with respiratory failure, pneumonia, cardiac arrest, and autonomic dysfunction being the most frequently described causes. Mortality risk factors include older age, severe presentation, and mechanical ventilation requirement. The leading causes of death during the progressive phase are mostly related to complications from autonomic dysfunction, while during the recovery phase, deaths are usually related to respiratory infections or cardiovascular complications10,20,23.
CONCLUSION
GBS is often associated with unfavorable functional prognosis in patients who are not diagnosed and treated timely. During the past decade, several advances have been made in understanding the pathophysiological drivers of nerve injury in GBS that may support the development of targeted therapies. However, prognostic biomarkers and targeted treatments are still needed. A clear link between COVID-19, the currently available vaccines against SARS-CoV-2, and GBS is yet to be established in large-scale epidemiological studies. Hence, physicians should be aware of the diverse clinical presentations, diagnostic, and care protocols of GBS.

