











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Pituitary adenomas (PA) are benign epithelial tumors arising from the endocrine cells of the anterior pituitary gland and comprise 10-25% of all intracranial neoplasms, third only to meningiomas and glioblastomas1. The prevalence of PA among autopsy and radiological studies is 16.7% and 22.5%, respectively2,3. The majority of these incidentally found lesions are microadenomas measuring < 10 mm in diameter2. The age-adjusted incidence rate of PA is estimated to be 3.4 cases per 100,000 individuals per year2. PA are very rare in children, with a prevalence of 0.1/million4. PA are broadly classified as functioning and non-functioning lesions, depending on whether or not they produce a hormonal hypersecretion syndrome2,5. The most frequent functioning PA are prolactinomas, followed by growth hormone (GH)-secreting somatotropinomas (causing acromegaly/gigantism), adrenocorticotropic hormone (ACTH)-secreting corticotropinomas (causing Cushing disease), and the less common TSH-secreting thyrotropinomas (causing central hyperthyroidism)2. In community-based studies, prolactinomas and clinically-nonfunctioning PA account for 47% and 33% of all PA, respectively5-8. The prevalence of clinically diagnosed PA is between 78 and 94 cases per 100,000 inhabitants5-8. Non-functioning PA are the most common type when considering only macroadenomas, whereas prolactinomas predominate when considering both micro- and macro-adenomas5-8.
More than 95% of PA occur sporadically, without a recognized familial or inherited cause9,10. X-chromosome inactivation and microsatellite analysis indicate that PA are monoclonal neoplasms11. Sometimes somatic changes occurring only in tumoral pituitary tissue are recognized as recurrent events and include GNAS (encoding the alpha subunit of the guanine nucleotide-binding protein) mutations in patients with acromegaly12 and USP8 (encoding ubiquitin carboxyl-terminal hydrolase 8) mutations in Cushing’s disease13. While these somatic genetic changes may confer some peculiar clinical features, such as increased responsiveness to therapy with somatostatin analogs14,15, they cannot be readily identified a priori. Indeed, most somatic genetic changes seen in PA subtypes are of uncertain pathogenic significance. This will likely change as a greater number of driver mutations causing pituitary tumorigenesis are identified. In 5% of PA, a number of inheritable genetic conditions have been identified at the germline level (9-10). These genetic or genomic disorders may be limited to PA alone or may be associated with involvement of other organs in a syndromic endocrine neoplasia setting. Hereditary pituitary tumors can be classified as syndromic (multiple endocrine neoplasia type 1 [MEN1], MEN4, Carney complex [CNC], familial paraganglioma-pheochromocytoma-PA syndrome, and McCune-Albright syndrome [MAS]) and isolated or non-syndromic (Familial Isolated PA and X-linked acrogigantism [XLAG])9-10.
Several factors are thought to be involved in the pathogenesis of pituitary tumors; these include genetic mutations, epigenetic dysregulation of cell cycle regulators, local growth factors, and possibly hypothalamic dysregulation16-18. Two key mechanisms are involved in the PA tumorigenic process: gain of function or activating mutations of oncogenes and loss of function or inactivation of tumor suppressor genes (TSG), which can occur either independently or in combination9-10,16-18.
In contrast to oncogene activation, TSG inactivation usually requires the loss of both alleles, according to Knudson’s “two-hit” hypothesis19. The first hit may be an inherited heterozygous germline mutation, a somatic mutation, or the loss of one allele of a particular TSG; as long as the other gene remains functional, the disease or trait is not expressed17-19. However, when the other allele is altered (the so-called second hit), usually by a partial chromosomal deletion or silenced by methylation of its promoter, the full phenotypic expression of the condition ensues17-19. Such partial chromosomal deletion may lead to loss of heterozygosity (LOH) of common polymorphisms around the TSG locus18. A recently described second hit mechanism involves the upregulation of a microRNA which then turns off the remaining TSG allele20. LOH has been described on chromosomes 9, 11q13, and 13 in approximately 20% of cases of sporadic PA9,10.
In this review, we aim to discuss the genetic causes and clinical spectrum of familial pituitary tumors, focusing on germline and somatic mosaic mutations causing familial isolated pituitary adenomas (FIPA) and syndromic conditions, including MEN1, MEN4, CNC, MAS, familial paraganglioma/pheochromocytoma/PA, and DICER1 syndrome (Table 1).
Table 1 Germline and mosaic mutations causing hereditary pituitary tumor syndromes
| Syndrome | Affected gene | Function | Chromosomal locus | Penetrance (%) | Clinical characteristics |
|---|---|---|---|---|---|
| MEN 1 | MEN1 | Tumor suppressor | 11q13.1 | 30-40% | Hyperparathyroidism, PAs (mostly prolactinomas and GH-secreting), NET, other neoplasms |
| MEN 4 | CKDN1B | Tumor suppressor | 12q13.1 | unknown | MEN-1 like, usually with GH-secreting pituitary adenomas |
| Carney Complex | PRKAR1A | Tumor suppressor | 17q24.2 | 10-15% | Skin pigmentation; cardiac and cutaneous myxomas; thyroid, testis and adrenal tumors, pituitary hyperplasia, and PAs |
| Pheochromocytoma/paraganglioma/pituitary adenoma syndrome |
SDHA
SDHB SDHC SDHD MAX |
Oncogene | 5p15.33 1p36.13 1q23.3 11q23.1 14q23.3 | <1% <1% <1% <1% Unknown | Familial pheochromocytomas and paragangliomas (PPGLs) |
| DICER 1 Syndrome | DICER1 | RNA interference | 14q32.12 | <1% | Early onset pituitary blastomas (adrenocorticotropic hormone secreting), pleuropulmonary blastoma, ovarian sex cord-stromal tumors, cystic nephroma, thyroid cancer |
| FIPA | AIP | Tumor suppressor | 11q13.2 | 15-30% | Young-onset somatotroph or mixed somatotroph-lactotroph PAs and prolactinomas. Responsible for 15-30% of FIPA kindreds and up to 20% of young-onset PAs, often resistant to pharmacological therapy |
| XLAG | GPR101 | Oncogene | Xq26.3 | 100% | Early-onset (< 4 years) gigantism |
| McCune-Albright syndrome | GNAS1 | Oncogene | 20q26.3 | 20% | Polyostotic fibrous dysplasia, café-au-lait spots, and precocious puberty with GH and/or PRL excess |
MEN1: multiple endocrine neoplasia type 1; GEP NET: gastroenteropancreatic neuroendocrine tumor; MEN4: multiple endocrine neoplasia type 4; NFPA: non-functioning pituitary adenoma; PA: pituitary adenoma; FIPA: familial isolated pituitary adenoma.
MEN1
MEN1 is a high-penetrance, autosomal dominant condition characterized by the development of parathyroid, pituitary, and pancreatic tumors21,22. Other, less common components of MEN1 are carcinoid tumors, adrenocortical tumors, facial angiofibromas, lipomatous tumors, and collagenomas21,22. MEN1 was formally described clinically in 1953 by Underhall et al.,23 and its autosomal dominant inheritance pattern was first suspected by Werner in 195424. It is a rare condition, with a 0.25% prevalence in autopsy studies, affecting 1 in 30,000 individuals21,22,25,26. The majority of MEN1 cases (90%) occur in a familial-hereditary setting, and only 10% occur sporadically21,22,25,26. MEN1 is the ultimate diagnosis in 1-18% of patients with primary hyperparathyroidism, 16-38% of patients with gastrinomas, and in < 3% of patients with pituitary tumors. The disease affects both females and males equally, and the age at presentation ranges between 5 and 81 years, with 95% of the patients developing the clinical manifestations by the fifth decade of life21,22,25,26.
Primary hyperparathyroidism is the most common clinical manifestation of MEN1 with a 95% penetrance25,26. Primary hyperparathyroidism occurring in the context of MEN1 has an earlier age of onset (20-25 years) than that occurring in non-MEN1 patients (50-55 years)25,26. Pancreatic islet cell tumors have a prevalence of 30-80% and include gastrinomas, insulinomas, glucagonomas, VIPomas (vasoactive intestinal peptide-secreting tumors), and somatostatinomas. MEN1 patients with pancreatic islet cell tumors on average have an earlier age of onset compared to non-MEN1 patients with similar lesions25,26. Gastrinomas are the most common pancreatic islet cell tumors in MEN1 patients, accounting for over half of all pancreatic islet cell tumors seen in these subjects25,26. The majority of gastrinomas occurring in the context of MEN1 are malignant25,26. Insulinomas account for nearly 10-30% of all pancreatic islet cell tumors and can coexist with gastrinomas in 10% of MEN1 patients25,26. Glucagonomas are seen in < 3% of MEN1 patients. The clinical features of glucagonoma include diarrhea, weight loss, anemia, necrolytic migratory erythema, and stomatitis25,26. PA occur in 15-90% of MEN1 patients, and the majority of these are prolactinomas (60%), followed by somatotropinomas (25%), clinically non-functioning PA (CNFPA) (10%), and corticotropinomas (5%)26,27. Patients with PA occurring in the context of MEN1 are younger and more frequently harbor macroadenomas than those occurring sporadically26,27.
Approximately 80% of patients with MEN1 harbor mutations of the MEN1 gene21,22. Although a few MEN1 cases are due to mutations in genes coding for other cell cycle regulators such as p15, p18, and p21, in 15-20% of cases no mutations can be found21,22. The MEN1 susceptibility gene was initially linked to a locus on chromosome 11q13 in 1988 by Larsson et al.28, and the MEN1 gene was subsequently cloned in 199729,30. The MEN1 gene has ten exons of which exons 2-10 encode a 61 aminoacid nuclear protein called, menin, whose functions are still being elucidated29,30. Menin appears to be located mostly in the nucleus, where it has multiple binding partners, including jun-D and members of the histone methyltransferase complex21,22. Menin potentially interacts with promoter regions of many genes, indicating its wide transcriptional regulatory role21,22. It has long been hypothesized that the tumor suppressive actions of menin are mediated through the regulation of histone methylation in promoters of p27 and Hox genes and possibly other cyclin-dependent kinase (CDK) inhibitors31.
About 75% of MEN1 mutations are inactivating, consistent with what would be expected for a TSG21,22,32. Somatic MEN1 mutations are commonly found in sporadic parathyroid (20%) and pancreatic neuroendocrine tumors (NET) (30%)21,22,32. These mutations are extremely rare in sporadic PA. LOH at 11q13 has been described in 30% of sporadic PA although MEN1 mRNA is not downregulated in these tumors21-23,33.
MEN4
Around 3% of patients with MEN1 have no identifiable menin mutations22,32,34. In some of these patients, abnormalities in genes encoding other CDKs have been identified22,32,34. These patients are classified as having MEN4 and the majority harbor germ-line, heterozygous, and non-sense mutations of CDKN1B (inhibitor 1B) that results in a truncated p27 protein34-41. CDKN1B is a TSG located on chromosome 12q13 in humans and encodes the CDK p27. CDKN1B transcription is regulated by menin, which enhances the activity of its promoter through interaction with histone methyltransferases31. p27 levels are also regulated through mitogen-activated protein kinase and phosphatidylinositol-4,5-bisphosphonate 3-kinase catalytic subunit42. Loss-of-function mutations in CDKN1B lead to decreased cellular levels of p27 and/or to p27 functional defects42. Until 2019, only 29 MEN4 cases were reported34. Very recently, Frederiksen et al. described a large Danish family with 13 cases of multiple endocrine tumors, none of which harbored any abnormalities in menin, that segregated with a pathogenic CDKN1B variant (c.121_122delTT, p.Leu41Asnfs*83)43. A comprehensive phenotype of MEN4 patients is not yet established due to the small number of patients identified so far. For the same reasons, the penetrance of MEN4, the frequency of familial and sporadic cases, and the phenotype-genotype correlations cannot be established yet. Primary hyperparathyroidism is present in all reported MEN4 patients34,42. Other MEN-1-like tumors identified are PA (corticotropinomas, somatotropinomas, and CNFPA), NET, adrenocortical tumors, and meningiomas as well as uterine neoplasms34,42.
CNC
CNC is a rare autosomal dominant condition with variable penetrance, characterized by various endocrine and non-endocrine abnormalities44,45. Approximately a total of 750 patients have been described to date44,45. Cardiac myxomas that could be localized in any of the heart cavities are reported in 30% of the patients and may result in heart failure44,45. Myxomas have also been found in breast and skin44,45. Lentigines are among the most common cutaneous manifestations of CNC and occur in 70% of the patients. These skin lesions are small brown or black macules located around the lips, on eyelids, ears, and genital area44,45. Other skin findings that can be seen in half of the patients include Spitz nevi, blue nevi, schwannomas, and café-au-lait spots44,45. The most common endocrine abnormality is an ACTH-independent Cushing’s syndrome, caused by micronodular pigmented adrenal hyperplasia, occurring in 25-30% of the patients44-46. Testicular, and to a lesser extent, thyroid nodules are relatively frequent44,45. Testicular nodules are usually Sertoli cell neoplasms, which may present as precocious puberty44,45. About 75% of patients with CNC exhibit asymptomatic elevations of both, GH and insulin-like growth factor 1 (IGF-1), usually associated with hyperplasia of the pituitary somatotroph; however, clinically evident acromegaly is rare48,49.
CNC is a genetically heterogeneous disease. Linkage analysis has identified three chromosomal loci to be associated with the disease: 17q22-24, 2p16, and 1p31.144-47. Over 70% of families with CNC harbor a germline-inactivating mutation of the alpha-1-regulatory subunit of the cAMP-dependent protein kinase A (PRKAR1A) gene, located on chromosome 17q2444-47. Although the causative gene has not been identified, in 18% of CNC families, the molecular defect has been localized to 2p1644,45,47. A single patient has recently been described with a duplication event in 1p31.144,45,47.
The PRKAR1A gene spans a 21-Kb region and consists of 11 exons, comprising a coding region of 1143 bp that encodes a 381-amino acid protein of 42.9 Kd45,47. In its inactive state, PKA is a tetrameric enzyme consisting of two regulatory and two catalytic subunits. Four isoforms of the regulatory subunit have been described: R1alpha (encoded by PRKAR1A), R1beta, R2alpha, and R2beta, which interact forming homo- and hetero-dimers. Similarly, the catalytic subunit forms dimers giving rise to four different isoforms: Calpha, Cbeta, Cgamma, and PRKX. cAMP is bound by regulatory subunits (R2[cAMP]4), thus dissociating the two catalytic subunits, rendering them free to phosphorylate and activate proteins such as cAMP response element-binding protein (p-CREB) that result in the transcription of genes encoding proteins which promote cell proliferation and inhibit apoptosis (Fig. 1). Phosphorylated CREB also increases the transcription rate of Pit-1 that promotes the differentiation of thyrotrophs, somatotrophs, and lactotrophs44,45 and the transcription of genes encoding TSH, GH, and prolactin. Thus, the main role of the regulatory subunits is to limit the action of the catalytic subunits45-47. Inactivating mutations of PRKAR1A result in an unrestricted action of the catalytic subunits and thus, promote cell proliferation and hormone production. Over 40 different mutations have been described, including point mutations, insertions, small deletions, and combined rearrangements of less than 15 bp44,45,47. The majority of these mutations generate a direct or frameshift premature stop codon, which results in a truncated protein. A two base-pair deletion in exon 5 of the gene is the most frequently seen mutation in CNC patients44,45,47. This genetic defect is found de novo in approximately 30% of CNC cases. Truncating PRKAR1A mutations result in mRNA instability due to a nonsense-mediated decay mechanism44,45,47. LOH at 17q22-24 and loss of the normal allele have been demonstrated in CNC tumors. PRKAR1A somatic mutations have not been identified in pituitary adenoma (PA) but have rarely been described in sporadic thyroid tumors50.
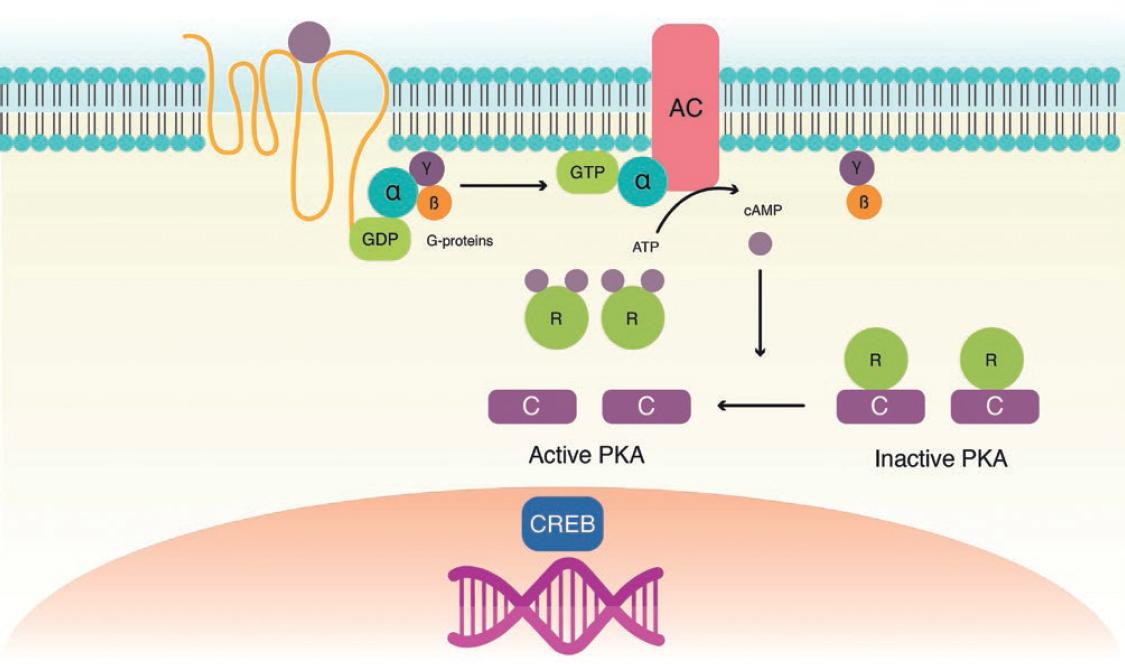
Figure 1 Molecular pathophysiology of carney complex. Receptor activation results in guanosine triphosphate (GTP) binding to the alpha subunit of the Gs-protein, dissociating it from the beta/gamma complex. The GTP-bound alpha subunit activates adenylate cyclase, leading to increased cAMP generation. cAMP activates protein kinase A by binding to its regulatory subunit (R), leaving the catalytic subunit (C) free for serine-threonine phosphorylation and activation of proteins such as cAMP response element-binding (CREB) protein. Inactivating mutations of the R subunit decrease its binding to the C subunit, which results in increased activation of CREB and other target proteins, leading to increased cellular proliferation, and inhibition of apoptosis.
Pheochromocytoma/paraganglioma (PPGL)/PA syndrome
Although the first description of coexisting PA and PPGL dates back to 195251, it was not until 2012 that a causal link between genes predisposing to catecholamine-secreting tumors and a GH-secreting PA was established52. In this report, Xekouki et al. describe a hypertensive 40-year-old male patient with acromegaly due to a large GH-secreting PA who also had bilateral pheochromocytomas and multiple paragangliomas52. Genetic testing revealed the presence of a succinate dehydrogenase-D gene (SDHD) germline, frameshift mutation leading to a premature stop codon, as well as LOH of the SDHD locus in the resected PA52. The same mutation was also found in two affected relatives, including a paternal uncle who had been operated many years before for bilateral neck masses occurring in the context of uncontrolled hypertension52. A total of 74 patients having both PPGL and PA have thus far been reported: about 22 (29.7%) with identified mutations in predisposition genes, 23 (31%) with a personal or family history suggestive of a hereditary endocrine syndrome, and 29 (39%) as isolated cases10,53.
The SDH mitochondrial complex consists of four subunits (A, B, C, and D), which form the catalytic enzymatic core (A and B) and anchor the complex to the inner mitochondrial membrane (subunits C and D), plus its associated assembly factor (SDHAF2)54-56. SDH is a key enzyme of the tricarboxylic acid and electron transport chain, catalyzing the conversion of succinate to fumarate54-56. Disruption of this enzymatic complex results in succinate accumulation which in turn inhibits prolyl hydroxylases; the inability to hydroxylate hypoxia-inducible factor 1a (HIF1a) leads to a pseudohypoxic state that promotes the transcription of HIF-responsive genes54-56.
Pituitary blastoma and DICER1 mutations
DICER1 syndrome, also known as pleuropulmonary blastoma (PPB)-familial tumor and dysplasia syndrome, is caused by a heterozygous germline mutation of the DICER1 gene. The first case of pituitary blastoma was described in 2008 in a 13-month-old female child who presented with Cushing’s disease and diabetes insipidus57. The main manifestations are PPB, cystic nephroma, Sertoli-Leydig cell tumors, goiter, and more rarely, sarcomas, dysplasias, and pituitary blastoma58,59. Characteristically, the presentation of the clinical components of this syndrome is age-specific58,59. Despite its low penetrance (< 1%), pituitary blastoma is considered a pathognomonic feature of DICER1 syndrome. The median age of presentation is 8 months (range 7-24 months)58,59. Symptoms of Cushing’s disease and ophthalmoplegia are the most common presenting manifestations of pituitary blastoma58,59.
The DICER1 gene, located on 14q32.13, encodes a small RNA processing endoribonuclease that cleaves precursor miRNAs into mature miRNAs, which in turn regulate mRNA expression60. The pathogenesis of pituitary blastoma encompasses the existence of a loss-of-function DICER1 germline mutation plus the occurrence of a second somatic hit, which is crucial for tumor development in the embryonic pituitary61.
ISOLATED PITUITARY ADENOMA
Familial isolated PA
The term familial isolated PA alludes to a syndrome whereby two or more cases of a PA are found within a family, without any clinical or genetic features of MEN1 or CNC9,10,62. This autosomal dominant condition with incomplete penetrance is also known as isolated familial somatotropinoma since GH-secreting adenomas are the most commonly found tumors. Since its description in 2006, several hundred families have been reported9,10,62.
The genetic defect in this syndrome was elucidated by linkage-disequilibrium analysis of a Finnish family with several members affected by GH and prolactin-secreting adenomas9,10,63. Whole-genome single nucleotide polymorphism genotyping of 11 affected individuals, found a haplotype involving 11q12-13 that perfectly segregated with the presence of acromegaly (LOD score 7.1) (63). The 11q12-13 locus encompasses over 60 Mb and more than 29 genes9,10. Comparative genomic hybridization using 172 probes found one gene to be associated in a statistically significant manner63. This gene is currently known as the AIP gene (AIP: aryl hydrocarbon receptor-interacting protein), but it was formerly known as ARA9 or XAP29,10,62-64.
The AIP gene is located on chromosome 11q13, 3 Mb distal to the MEN1 gene62,64. It contains six exons and encodes a 330 amino acid co-chaperone protein which is well-conserved throughout evolution62,64. The amino-terminus of the AIP protein has an immunophilin-like domain, with significant homology to immunophilins FKBP12 and FKBP52 (Peptidyl-prolyl cis-trans isomerase)62,64. However, it differs from other immunophilins by not sharing the ability to bind to immunosuppressant drugs such as cyclosporin or rapamycin62-64. The carboxy-terminus contains seven alpha helices that are crucial for protein-protein interactions: three 34 amino tetratricopeptide (TPR) domains, each with two helices, and a final seventh alpha helix62-64. The function of AIP is to stabilize the aryl hydrocarbon receptor/heat shock protein-90/dioxin complex in the cytoplasm, preventing it from being translocated into the nucleus62-64.
Over 100 different AIP mutations have been identified to date, including deletions, insertions, segmental duplications, nonsense, and missense mutations as well as deletions of whole exons or even the whole gene62,65. Most of the pathogenic missense mutations directly affect the TPR domains or the C-terminal alpha-helix62,64,65. Two-thirds of the AIP mutations lead to protein truncations, which remove segments of the TPR domains and/or carboxy-terminal end, and therefore, lead to loss of function of the protein62,64,65. A common genetic “hotspot” for mutations in the AIP protein is the 304 residue (R304X and R304Q), which affects a CpG sequence and has been shown to be present in several independent families from different parts of the world65-68. Other potential hotspots include the 271 and the 81 loci (65-66). Inactivating AIP mutations are responsible for 20% of FIPA cases62,65. Germline, heterozygous AIP mutations are also present in 5-10% of cases of sporadic GH-secreting adenomas65-67. Acromegaly patients with AIP mutations are usually diagnosed before age 30, usually harbor macroadenomas, frequently cosecrete prolactin and appear to be somewhat less responsive to treatment with somatostatin analogs65-67.
In AIP mutation-negative families, the pituitary tumors are also predominantly macroadenomas (71%)65,66. The observed tumor types are also dominated by prolactin and GH-secreting tumors, but CNFPA and rarely corticotroph adenomas have also been described65,66. The male-to-female ratio is 1:1, and the age of onset is more similar to sporadic PA patients65,66. Penetrance is probably slightly lower than in AIP-positive families65,66.
A recent study has demonstrated that upregulation of AIP in the liver of transgenic mice increases the expression of the TSG ZAC1 (zinc finger regulator of apoptosis and cell cycle arrest)69,70. This is likely the mechanism by which AIP exerts its tumor suppressor effects in the pituitary. A recent study demonstrated that ZAC1 mRNA expression was significantly increased in GH3 cells transiently transfected with wild- type AIP compared to the empty vector and those transfected with mutant forms of AIP (C238Y, and R304X)69,70. New data from in vitro experiments on mouse embryonic fibroblast and PA cell lines demonstrate that AIP deficiency results in increased cAMP levels through defective Gai signaling71. This results in subsequent downregulation of phosphorylated extracellular signal-regulated kinases 1/2 (p-ERK1/2) and p-CREB71. This new evidence suggests that defective Gai signaling is potentially a major contributor to the development of GH-secreting PA in AIP mutation carriers71.
X-linked acrogigantism
X-linked acrogigantism (XLAG) is a recently described condition characterized by severely increased linear growth developing at a very early age. This is very rare form of gigantism, first described in 2014 by Trivellin et al.72, results from microduplications of Xq23.6, encompassing a 500-bp sequence that includes the GPR101 gene73. Germline and to a lesser extent, mosaic mutations have been described, occurring both, in a familiar and sporadic setting72-77. Only 33 XLAG patients have been reported in the medical literature72-77. XLAG accounts for 8-10% of all gigantism cases76,77. In contrast to gigantism due to AIP mutations whereby the majority of patients are male, in XLAG, there is a clear female predominance72-77. The principal manifestation is increased linear growth starting at a very early age72-77. Patients with XLAG have normal weight and height at birth and develop accelerated growth usually before the age of 572-77. Other clinical features include acral enlargement, coarsened facial features, increased appetite, acanthosis nigricans, sleep apnea, and hyperhidrosis72-77. Biochemically, patients with this condition show very high basal and glucose-suppressed GH levels, as well as elevated age-adjusted IGF-1 concentrations; 85% have hyperprolactinemia72-77. Patients with XLAG usually have somatotroph hyperplasia and less commonly, well-defined adenomas73,76,78. Reticulin staining shows the characteristically distorted pattern of PA, and in some cases pseudofollicles and an eosinophilic colloid-like material are found. GH, PRL, and Pit-1 immunostaining are intense and diffuse73,76,78. Cam 5.2 immunostaining reveals the typical nuclear fibrous bodies of sparsely-granulated adenomas63,64,73,76,78.
The GPR101 gene encodes a Gs-protein-coupled orphan receptor that is normally expressed in the nucleus accumbens, medulla, and occipital cortex and abundantly expressed in pituitary tumors of XLAG patients79. The oncogenic processes resulting from GPR101 duplication are not completely understood; however, constitutive activation of this receptor could trigger the cAMP-PKA pathway and thus, cellular proliferation and autonomous GH hypersecretion of somatotrophs78. Since GPR101 appears to be involved in the regulation of GHRH release, hypersecretion of this hypothalamic hormone may contribute to somatotroph hyperplasia and increased GH secretion78.
MOSAIC MUTATIONS
McCune-Albright
McCune-Albright (MAS) was first described in 1937 by Donavan James McCune and Fuller Albright80,81. This is a rare and sporadic condition resulting from postzygotic mosaic mutations in the gene encoding the alpha subunit of the Gs protein (GNAS)82,83. Diagnosis of MAS is established on clinical grounds with patients having at least two features of the triad of polyostotic fibrous dysplasia (FD), café-au-lait skin pigmentation, and autonomous endocrine hyperfunction, including precocious puberty, thyrotoxicosis, pituitary gigantism, and Cushing syndrome as well as renal phosphate wasting49,84. The most common forms of autonomous endocrine hyperfunction in this syndrome are gonadotropin-independent precocious puberty, followed by thyrotoxicosis, pituitary gigantism, and Cushing syndrome. Café-au-lait spots are commonly the first manifestation and usually appear at birth or shortly thereafter. However, it is most often precocious puberty or FD that bring patients to medical attention49,84. FD is characterized by the lack of differentiation and proliferation of bone-forming stromal cells leading to replacement of normal bone and marrow by fibrous tissue49,84. FD most commonly behaves as a slowly growing mass lesion. Symptoms and signs vary depending on the type and location of FD and include craniofacial deformity, visual and hearing impairment, nasal congestion and/or obstruction, paresthesia, and pain49,84. The most commonly involved areas are the proximal femur, the craniofacial bones, and the ribs49,84.
Pituitary disease in MAS consists of lactotroph and somatotroph cell hyperplasia or adenomas, resulting in prolactin and GH excess, respectively85,86. In up to 20% of patients with MAS, there is a lack of GH suppression on glucose loading85,86. The elevated GH and IGF-1 levels can result in acromegaly/gigantism and have been implicated in the sarcomatous transformation of osseous FD86,87. Clear-cut pituitary tumors can be found in only 50% of patients. Diagnosis of GH excess can be challenging in MAS patients. In children with MAS, rapid linear growth which could be a result of GH excess is often attributed to precocious puberty, which is a common finding in patients with MAS85,86. In addition, characteristic features of acromegaly such as coarsening of the face, frontal bossing, and prognathism not only develop insidiously but can also be wrongly attributed to FD of the skull which can result in dysmorphic features85,86.
GNAS is located on chromosome 20q13 and encodes the ubiquitously expressed stimulatory Gs alpha subunit of the G protein that has intrinsic GTPase activity86-88. Missense mutations at codons 201 or 227, result in loss of GTPase activity and lead to constitutive and permanent activation of adenylate cyclase and hence, of the cAMP-dependent PKA pathway82,83,87-89. MAS is due to early postzygotic GNAS mutations occurring at codon 201 resulting in somatic mosaicism, which underlies the complex phenotype of these patients81,82,86,88. A variable proportion of sporadic somatotropinomas harbor somatic mutations of either codon 201 or 227, that result in constitutive activation and thus, in increased cellular proliferation and unrestricted GH secretion12,14,89,90. The GNAS1 gene is a paternally imprinted gene in many tissues, including the pituitary gland; therefore, only mutations occurring on the maternal allele will result in an abnormal phenotype91. Whereas up to 40% of Caucasian patients with acromegaly harbor GNAS1 mutations12,14,89,90, this occurs in only 5-10% of Asian patients92,93 and 15-20% of Latin-American Mestizo patients15,94. Acromegaly patients with GNAS1 mutations are likely to have a relatively milder phenotype with smaller, less invasive, usually densely-granulated adenomas that seem to respond better to surgery and treatment with somatostatin analogs14,90,94.
Familial hyperprolactinemia
A heterozygous mutation of the prolactin receptor has recently been described in members of a family with mild to moderate hyperprolactinemia and normal pituitary gland on MRI95. Some family members have fertility problems, while others are completely asymptomatic. In vitro expression studies have shown that this prolactin receptor mutant is not only uncapable of properly transducing a signal but also exerts a dominant-negative effect95.
CONCLUSIONS
Although < 5% of PA occur in the context of well-defined genetic syndromes, their molecular pathophysiology has helped us understand the potential mechanisms involved in the oncogenesis of sporadic PA in general. Some of these molecular abnormalities occurring in the germline in these hereditary syndromes can be found as somatic mutations in sporadic PA. PA found in patients with hereditary syndromes are usually larger, more invasive, and less responsive to pharmacological treatment. At present, genetic testing looking for abnormalities in GNAS, Menin or PRKAR1A is recommended in patients with apparently sporadic PA who have a family history of pituitary tumors as well as in those who are relatively young at the time of diagnosis (less than age 30).

