











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Primary central nervous system lymphoma (PCNSL) is a rare variant of extranodal non-Hodgkin lymphomas, with a reported annual incidence rate of 4 cases per 106 adults. pcnsl account for 2.4-3% of all primary central nervous system (CNS) tumors, with 30-40% of diagnosed cases consisting of diffuse large b-cell lymphoma (DLBCL).1,2 Patients immunocompromised remain the only well-identified group at increased risk.3 The clinical presentation varies widely with sub-acute and non-specific onset, conditioned by the brain area affected. Distinctively, immunocompetent patients experienced focal encephalic, spinal, leptomeningeal, or eye lesions sans evidence of systemic disease.4 In the setting of neurofibromatosis type 1 (NF-1) patients, most malignant lesions are markedly associated, predominated by gliomas and malignant peripheral nerve sheath tumors (mpnst) with an average age of diagnosis between 12-27 and 33 years accordingly.5 However, B-Cell lymphoma has been reported as a poorly related entity to NF-1, decreasing the index of suspicious malignancies during follow-up.6,7 We describe a pcnsl as the first evidence of a malignant lesion in a patient initially diagnosed with NF-1. To our knowledge, this is the third report detailing a singular coexistence between these two conditions.
Case presentation
Patient history
A 53-year-old female with NF-1 presented with worsening progressive motor symptoms. She had initially developed sudden difficulty with fine motor tasks a month before the medical visit. Our patient specified a particular straining to grasp pens and cups. In this occasion, her symptoms reached their plateau with rest. One week prior to consultation, the patient experienced severe headaches with secondary features, vas 8/10, associated with nausea and no medical relief with analgesics. Afterward, she presented to the emergency room with subacute and progressive left-sided numbness and weakness.
Clinical examination demonstrated left hemiplegia (MRC 2/5), associating the progression to an incomplete pyramidal syndrome.
Neuroimaging and surgical approach
Magnetic resonance imaging (MRI) showed a left central lobe, heterogenous, gadolinium-enhanced mass with perilesional edema, 30 mm in diameter (Figure 1). 19-fdg pet scan provided additional information about activity exclusively in the CNS. Once identified, the mass was surgically resected by craniectomy with an interhemispheric approach and endoport- assisted surgical evacuation. We identified a highly vascular, round-shaped specimen intraoperatively with a yellowish appearance. There were no postoperative complications.
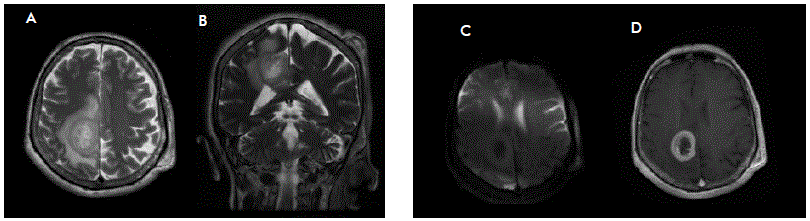
Figure 1 Magnetic resonance imaging. A-D Brain lesion identified during diagnostic assessment. Axial T2-weighted imaging (A) shows an intrinsic heterogeneously lesion located in the left central lobe, well delimited with significant perilesional edema. Contrast-enhanced T1-weighted imaging (D) demonstrates peripheral contrast uptake, with effacement of surrounding brain sulci. Coronal T2-DWI (B) shows no apparent restriction.
Histopathological findings
Atypical ovoid-shaped cells were observed, with scant cytoplasm and hyperchromatic nuclei, consistent with lymphoid lineage cells within Virchow-Robin space (Figure 2).
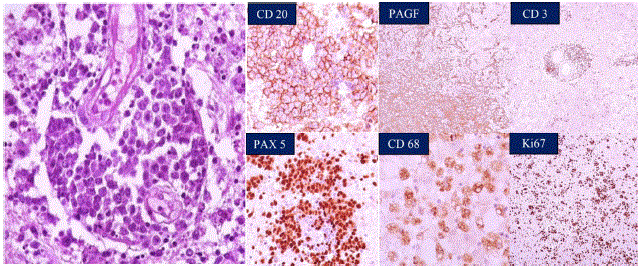
Figure 2 Immunohistochemical stains from the brain mass biopsy. Lymphoid lineage cells within Virchow-Robin space are shown, with atypical ovoid- shape, scant cytoplasm, and hyperchromatic nuclei. Most cells in the specimen were CD20-positive B cells. There are background smaller CD3-positive T-cells. PAX-5 and Ki-67 were positive as well, highlighting neoplastic cells. Overall, the findings are most consistent with a diffuse large B cell lymphoma.
There were apoptotic bodies amid these spaces. Immunohistochemical staining was positive for CD20, PAX- 5, and Ki-67, highlighting neoplastic cells; there were CD3 stained in T cells.
Discussion
PCNSL remains an unduly rare subgroup of malignancies with unusual clinical course, aggressive behavior, and poor survival outcomes compared with other lymphomas. Current treatment has increasingly improved in the last two decades - associated with the advent of better therapeutic strategies - but has limited impact on survival rates. Front-line therapy consists of high-dose methotrexate- based poly-chemotherapy;8,9 surgery remains controversial considering no direct benefit to prognosis. However, small observational studies supported these results and are discordant to modern neurosurgery techniques reflected in contemporary studies.10,11 Due to specific patterns of this neoplasm, it is mandatory to reduce the gap between diagnostic suspicion and early therapeutic intervention, expressly in high-risk patient groups.
Of interest, our patient appertains to a high-risk population with a history of NF-1. Previous studies have identified NF-1 as a significant risk factor for malignancy,5 conversely, there is evidence against diffuse B cell lymphoma association with NF-1.12 This evidence undoubtedly contributes to and ratifies the need for a greater understanding of NF- 1-associated malignancies. The pathogenesis of PCNSL is still unclear, with some genetic observations hypothesizing a possible pathophysiological relationship with NF-1.13,14
NF-1 originates from mutations in the NF-1 tumor suppressor gene on chromosome 17q11.2, causing loss of production or function. This gene produces the GTPase-activating protein neurofibromin, which stimulates signaling pathways such as the mammalian target of rapamycin (mTOR), mitogen-activated protein kinase (MAPK), and stem cell factor (SCF), through the RAS p21 family.15 However, the risk of developing hematopoietic alterations of the lymphoid lineage in adults is not well investigated. Landry et al. studied a cohort of 1607 patients with NF-1. Different odds ratios estimated the risk for cancer related NF-1 versus the control group.5 Non-Hodgkin Lymphoma (NHL) reported an or of 0.8, with a mean age of 48.9 at diagnosis, which confirms a marked association between NF-1 and neoplasms compared to the general population. The pediatric population reported a high risk for acute lymphoid leukemia (all), and non-Hodgkin lymphoma (NHL).16 Molecular mechanisms involved in the pathogenesis of this type of cancer, especially in the setting of NF-1 adults, have not been identified. Hematopoietic neoplasms, astrocytomas, and MPNST were associated with cooperative genetic alterations, such as the biallelic inactivation of NF-1 and the mutation of TP53.5 Consequently, there is a hypothetical spectrum of NF-1 contributing to malignancies, such as this case of PCNSL.
Since these lymphomas primarily originate within the brain, they can involve the brain tissue, leptomeninges, eyeball, and spinal cord, without evidence of systemic disease. Most patients exhibit focal neurological deficits associated with mass effect and tumor location.1 Lesions are typically symmetric, located in the deep periventricular white matter, with occasional subependymal extension. The frontal lobe is primarily affected in 40% of cases, with a lower incidence presumably in the cerebellum and brain stem.17 Magnetic Resonance Imaging (MRI) T1 sequences frequently show hypointense or isointense lesions, while T2 shows isointense or hyperintense lesions. Lesions show moderate to high contrast-uptake and perilesional edema restricted to diffusion.4 Hence, glioblastoma multiforme fits among the differential diagnoses. In this case, the presumptive diagnosis was initially GBM, corresponding to its high incidence in NF-1 patients compared to purely NHL.5 The rationale supported surgical management and subsequently maximal safe resection with no further complications. Despite the selected therapeutic approach, histopathology assessment confirmed the diagnosis.
Much remains to be addressed in our understanding and management of CNS tumor variants among NF-1 patients. Despite multiple proposed theories for its pathogenesis, non-Hodgkin extranodal lymphomas as a primary manifestation within the NF-1 spectrum are still not fully comprehended and have an underestimated incidence. Recognition of key symptoms is often difficult, given the nonspecific nature of presenting symptoms. Once diagnosed, clear management strategies and contemporary surgical outcomes remain somewhat elusive. Therefore, reporting such cases is a key for clinicians’ awareness to enforce a high index of suspicion for malignancies during the follow-up of patients with a history of NF-1 beyond gliomas.
Conclusions
PCNSL is a rare form of non-Hodgkin lymphoma with a sub-acute and non-specific onset that mimic other brain tumors. Early suspicion and diagnosis with MRI and other advanced imaging techniques, especially FDG-PET, are critical in preventing progression. We emphasize the importance of this case to reach a correct diagnosis when presented with a similar scenario, with timely and effective treatment offered to the patient. Additionally, we encourage the study of this entity since its real incidence may be underestimated, and its genetic and pathophysiological mechanisms remain uncleared.

