











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Lenalidomide (IUPAC 3-(7-amino-3-oxo-1H-isoindol-2-yl)piperidine-2,6-dione), a thalidomide analog, is an immunomodulatory (IMiD) and antineoplastic agent used in multiple myeloma therapy1. Multiple myeloma is a malignant B-cell neoplasia characterized by the excess of monotypic plasma cells in the bone marrow. Established for the 1st time as agents with antiangiogenic properties, thalidomide, and the other IMiDs inhibit the production of interleukin, which is a growth factor for the proliferation of myeloma cells. In addition, they activate apoptotic pathways through cellular death by means of caspases 82.
In 2005, the Food and Drug Administration (FDA) approved the marketing of 5 mg and 10 mg Revlimid® (Lenalidomide) capsules for the treatment of transfusion-depending anemia patients due to low-risk myelodysplastic syndrome (MDS) associated to 5q deletion abnormality with or without additional cytogenetic abnormalities and multiple myeloma in combination with dexamethasone3,4. The European Medicines Agency issued the authorization to market lenalidomide in 20075. In Brazil, ANVISA issued a public opinion for the approval of Revlimid in 2018 for the treatment of patients with relapsed/refractory multiple myeloma who had received at least one prior treatment; and for the treatment of patients with transfusion-depending anemia derived from low to intermediate-1 risk MDS associated to 5q deletion cytogenetic abnormality, with or without additional cytogenetic abnormalities6.
In 2017, ANVISA issued resolution RDC 191 (regarding the control of lenalidomide and any drugs containing it, among other provisions), later revoked in 2022 by a resolution with the same name (RDC 735/2022). Due to the risks associated to the teratogenic effects of lenalidomide-based drugs, the resolution stated, among other provisions, the design of a Risk Management Plan, including a pregnancy prevention program, which requires training of the physicians prescribing the drug by the register holder7.
In 2020, ANVISA issued RDC 393/2020, which included lenalidomide therapeutic indications for the treatment of multiple myeloma, in combination with bortezomib and dexamethasone, for patients with no prior treatment; for the treatment of follicular lymphoma or previously treated patients with marginal zone lymphoma, in combination with rituximab (anti-CD20 antibody), and relapsed/refractory mantle cell lymphoma8.
As to the security profile, the authors report that lenalidomide is well tolerated and that most usual adverse events include hematologic toxicity with controllable neutropenia and thrombocytopenia9,10. According to the panel released by ANVISA through VigiMed, in the period between January 01, 2018, and September 09, 2022, 652 lenalidomide suspected adverse events were informed, out of which 16.10% corresponded to neutropenia and 4.45% to thrombocytopenia11.
The objective of this trial was to verify whether the rate and extent of absorption of the 25 mg lenalidomide immediate release capsule formulation manufactured by Eurofarma Laboratórios S.A. are equivalent to those of the reference product, REVLIMID® when administered in one single dose and under fasting conditions in adult healthy male subjects.
Methods
Study protocol and reporting
Bioequivalence studies are a type of study that follows specific rules dictated by the regulatory agencies in each country. Thus, study protocol drafting requirements follow the SPIRIT Statement12-14, where applicable. Data reporting is also carried out in the form of standardized reports by the agencies, but they contain all the information recommended for reporting according to the CONSORT Statement14,15. In Brazil, the following standards must be followed: (1) RE 894/2316, which defines the standardization for the preparation of bioequivalence study protocols, and (2) RE 895/2317, which defines the form of study reporting.
Study formulations
The test drug −25 mg lenalidomide immediate release capsule−, was manufactured by Eurofarma Laboratórios S/A. The reference product used in the study was Revlimid® (25 mg lenalidomide immediate release capsule), manufactured by Celgene Europe B.V. and registered in Brazil by Bristol-Myers Squibb Farmacêutica LTDA.
Study volunteers
Adult male healthy volunteers willing to participate in the study were selected based on the protocol eligibility criteria 90 days before the first study period. A sufficient number of eligible volunteers appeared in the research center facilities, and the 32 volunteers who fulfilled the protocol requirements were given information regarding the study, after having their inquiries clarified and having decided to willingly take part in the study, each subject signed the informed consent form, previously approved by the Avant Santé Research Center Research Committee (COFEPRIS 18 CI 19 019 021) along with the study protocol. Initially, 32 study subjects were selected and randomized, out of which only 28 completed all the study procedures due to exclusion and/or dropout reasons.
Study design
Single dose, randomized, open-label, two-treatment, two-sequence, two-period crossover bioequivalence study of 25 mg lenalidomide capsules manufactured by Eurofarma Laboratórios S.A. versus REVLIMID® (25 mg lenalidomide immediate release capsule) manufactured by Celgene Europe B.V. and registered in Brazil by Bristol-Myers Squibb Farmacêutica LTDA in adult male healthy volunteers under fasting conditions.
Drug administration
Study subjects were kept in fasting conditions for 10 h before the dose administration and at least 4 h afterward, on each period. The study was conducted under fasting conditions as required by ANVISA - Agência Nacional de Vigilância Sanitária18.
On each period of the study, a single dose (25 mg) of the test or the reference product was administered orally to the volunteers, in a seated position, with 200 mL of water at room temperature, under fasting conditions. The washout period was 7 days.
Blood sampling
A total number of 19 blood samples were collected from each volunteer in each period in tubes with K2EDTA. Blood samples (4 mL each) were collected at time points 0.00 h (before dose) and after administration at time points 0.25, 0.33, 0.50, 0.75, 1.00, 1.25, 1.50, 1.75, 2.00, 2.50, 3.00, 4.00, 6.00, 8.00, 10.00, 12.00, 16.00, and 24.00 h.
Biological samples processing
After collection, blood samples were processed in a refrigerated centrifuge at 3000 rpm for 10 min at 4 ± 2°C. The plasma obtained from the blood samples was transferred to two different cryogenic tubes (aliquot 1 and aliquot 2) previously identified and stored at a temperature below −50°C.
Lenalidomide quantification in plasma
METHOD VALIDATION
The validation of the bioanalytical method for quantification of lenalidomide in human plasma using Lenalidomide-d5 as internal standard and K2EDTA as anticoagulant through extraction in solid phase (Strata-X® 33 μm cartridge) and ultra-performance liquid chromatography-tandem mass spectrometry (LC-MS/MS) was performed in compliance with the acceptance criteria for selectivity, calibration curve, precision, accuracy, residual effect, matrix effect, and stability test in solution and the biological matrix (Table 1).
Tabla 1 Summary of the bioanalytical method
| Analyte | Lenalidomide |
|---|---|
| Internal standard | Lenalidomide-d5 |
| Biological matrix | Human Plasma |
| Anticoagulant | EDTA |
| Linearity | 2.044 ng/mL-1015.506 ng/mL |
| Curve equation | y = a + bx [1/x] |
| Lower quantification limit | 2.044 ng/mL |
| Low quality control | 5.148 ng/mL |
| Medium quality control | 411.867 ng/mL |
| High quality control | 792.052 ng/mL |
| Post-processing stability time | 2 days 8 h 38 min |
| Freeze/thaw cycles | 4 cycles |
| Short-term stability time | 6 h 18 min |
| Long-term stability time | 106 days |
EDTA: ethylenediaminetetraacetic acid.
Chromatographic conditions adopted for the validation and quantification of the study subject samples included the use of a Luna omega 1.6 μm PS C18 50 × 2.1 mm chromatographic column, at 35 ± 2°C. Samples were kept at 5 ± 4°C. Mobile phase A used was Formic Acid 0.1% and mobile phase B was LC-MS grade Acetonitrile in an 87:13 v/v proportion. The injection volume was 3.0 μL and retention times were 1.07 ± 0.3 min for the analyte and 1.05 ± 0.3 for the internal standard, the running time being 2.10 min.
The method proved linear between concentrations of 2.044 ng/mL to 1015.506 ng/mL according to equation y = a + bx [1/x], where "y" is the response, "x" is the analyte concentration and "1/x" is the selected weight.
The lower limit of quantification (LLQ) established for the method was 2.044 ng/mL and validated quality control samples were 5.148 ng/mL, 411.867 ng/mL, and 792.052 ng/mL.
STABILITY
Stability analysis was carried out in plasma in concentrations of 5.148 ng/mL and 792.052 ng/mL and they complied with the acceptance criteria when the samples were subjected to 6 h and 18 min at room temperature (approximately 25°C) (short-term stability), for 2 days 8 h and 38 min in auto-injector (5 ± 4°C) after sample extraction completion (post-processing stability) 4 freeze-and-thaw cycles and 106 long-term days.
STANDARD SOLUTIONS
Reference standards: lenalidomide (TRC/Canada) was used as an analyte and Lenalidomide-d5 (TRC/Canada) was used as an internal standard for the preparation of the primary standard solutions in methanol HPLC grade. Working solutions were prepared using milli-Q water: methanol (20:80; v/v) as eluent. All solutions were stored at a temperature between 2 and 8°C.
Compounds quantification in biological samples
Compounds were extracted from human plasma samples and quantified through LC-MS/MS using the Xevo TQ-S/Acquity UPLC I-Class (Waters) spectrometer, equipped with positive electrospray (ESP+) ionization source, and the analyte and internal standard were detected using multiple reaction monitoring with m/z transitions 260.21 > 149.11 and 265.23 > 151.13, respectively.
Results
Study population
The study began with 32 volunteers and ended with 28 healthy male volunteers between 18 and 42 years of age, who complied with the inclusion and exclusion criteria set forth in the protocol.
Pharmacokinetics and statistical analysis
Pharmacokinetic parameters Cmax and AUC0-t were established using software Phoenix WinNonlin version 8.3 and Sistema de Análise Estatística (SAS®) version 9.4.
Pharmacokinetic parameters are shown in table 2.
Tabla 2 Pharmacokinetic parameters (n = 28)
| Ratio (test/reference) | Geometric mean (%) | CI (90%) | Test power (%) | p (sequence) |
|---|---|---|---|---|
| Cmax (ng/mL) | 101.4 | 90.03-114.13 | 92.8 | 0.5299 |
| AUC0-t (ng.h.mL-1) | 94.4 | 88.21-101.02 | 100.0 | 0.1203 |
CI: confidence interval.
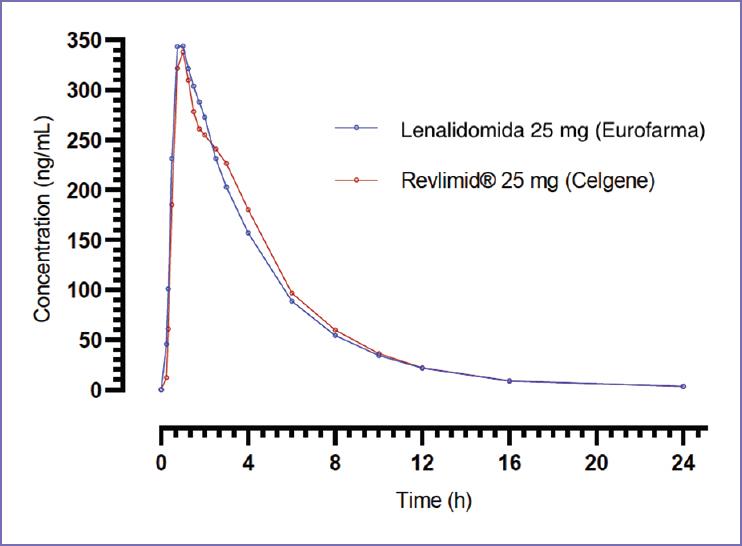
The maximum concentration of Cmax obtained for the reference product Revlimid® was 439.761 ng/mL in 1.5 h. For the test product, lenalidomide, a Cmax of 477.742 ng/mL occurred at 1.1 h. Figure 1 shows intermediate concentrations of lenalidomide for the 28 study subjects along collection times.
Tolerability/safety analysis
Eight adverse events were reported during the study: 3 headaches, 1 erythema, 1 rash, 1 hypertriglyceridemia, 1 rectal hemorrhage, and 1 normochromic normocytic anemia. No serious adverse events were reported; 6 events were classified as mild and 2 as moderate.
As to the relation with the study drug, 1 adverse event was classified as possibly related, 2 as probably related, and 5 as unlikely to be related to the study medication.
Discussion
The study was properly planned and conducted, obtaining pharmacokinetic parameters Cmax and AUC0-t which confidence interval values (90%) are within the acceptable limit for the ratio between the geometric means of the test and reference products (80−125%), according to the national legislation, ANVISA resolution RE no 1170/200619.
The planned number of 32 healthy male subjects is consistent with studies published by other authors20,21. The population selected for the study was male gender only, as recommended by the FDA guidelines22 and consistent with resolution ANVISA RDC 735/20227.
Both formulations were well tolerated during the study and no serious adverse events were reported. Headache was the most frequent adverse event, contrary to the reports of other authors and the ANVISA panel, who stated neutropenia and thrombocytopenia as the most frequent adverse events9,11,23.
The 7-day washout period seemed adequate, as all base collection samples of the volunteers of the second period had a concentration below the LLQ.
As in other published works, the analytical technique selected for the study of lenalidomide quantification of in human plasma samples was LC-MS/MS24-26.
Lenalidomide was quantified in its unaltered form, as required by the national legislation27.
Both reference and test drugs showed a maximum plasma concentration Cmax of 439.761 ng/mL and 477.742 ng/mL, respectively, consistent with those found in the literature20,21.
Conclusion
The results show test and reference formulations are considered bioequivalent regarding the absorption rate and extent, as the criteria required by the Brazilian regulatory authority have been complied with (CI 90% between 80 and 125%). Consequently, the test formulation, i.e. lenalidomide manufactured by Eurofarma Laboratórios S/A and the reference product Revlimid®, both in 25 mg capsules, are bioequivalent and therefore, interchangeable.
Authors contributions
C. Sverdloff participated in the study design and supervision. V. Marcondes Rezende performed the data curation and wrote the manuscript and reviews. L. de Cassia Val and L. Nerath Bonanato participated in the Project Administration and supervision. M.E. Cedano Limón conducted the clinical investigation. S. Kakarla performed bioanalytical method validation and supervision of sample analysis. M.H. Badii performed the statistical analysis. M. Pendela performed general site supervision. N.C. Lemus Castro wrote the clinical protocol, supervised clinical investigation, and was the principal investigator. All authors contributed with data and revision and approved the final manuscript.

